Population Genetics: Global clues to the nature of genomic mutations in humans
An analysis of worldwide human genetic variation reveals the footprints of ancient changes in genomic mutation processes.
- University of Cambridge, United Kingdom
All children inherit a mixture of chromosomal sequences from their parents, and although the copying process involved is extremely accurate, some errors occur. We refer to such errors as de novo (new) germline mutations, and when they are passed on to subsequent generations, these mutations are the raw material on which natural selection works and the source of all genetic differences between populations and species. Most have negligible or minor effects, but some on very rare occasions are responsible for serious genetic disease (Veltman and Brunner, 2012). The accumulation of genetic differences through mutation is also a primary source of information about human evolution. Thus it is important to understand the nature of genomic mutation, the rate at which it occurs and the factors causing it.
A key question is to what extent mutation processes differ between individuals, either in the total number of de novo mutations bequeathed to offspring or the places in the genome where they occur. Such differences could be genetic in origin, given that the proteins involved in DNA replication are themselves encoded in the genome, and there could also be environmental effects associated with where and how an individual lives. In both cases these factors may reflect recent evolutionary events, particularly the divergence of human populations and their global dispersal within the last 100,000 years.
One way to address this question would be to collect genomic and other data for thousands of families, identify de novo mutations in each of the offspring, and then analyse the factors contributing to them. However, this approach is extremely demanding in terms of the resources needed. Now, in eLife, Kelley Harris and Jonathan Pritchard of Stanford University report how they have taken an alternative approach (Harris and Pritchard, 2017) that involved using a dataset of whole-genome sequences for 2504 individuals from 26 different populations around the world (Auton et al., 2015). Consider that any genetic variant in this dataset, even if it is found today in many individuals, was once a de novo mutation in a single ancestor. Thus if different genetic or environmental factors have affected mutation processes in different human populations, we might expect to find evidence in the distribution of variants in these populations today.
Harris and Pritchard categorised single-nucleotide variants in this dataset by their ancestral and derived alleles (i.e. the version before and after mutation) and their sequence context as represented by the two flanking nucleotides. For example, the category AGC → ATC represents a mutation from G to T with flanking nucleotides A and C. After counting the number of variants in each category within every individual, the researchers found that the distribution of counts, termed the mutation spectrum, differs between populations to the extent that it is possible to identify an individual’s continent of origin based solely on the spectrum of mutations they carry. In general the differences between spectra comprise a multitude of small discrepancies, rather than large discrepancies in a few categories. However some categories do stand out, most notably an increased abundance of TCC → TTC mutations in European and South Asian populations – a signal also seen in other recent studies based on similar data (Mathieson and Reich, 2017; Narasimhan et al., 2016).
Harris and Pritchard then looked at how this signal changes with time. Mutations themselves have no timestamp, but on average a mutation that is rare in the population is likely to have arisen more recently than a mutation that is common. Using frequency as a proxy for age, Harris and Pritchard found that the enrichment for TCC → TTC was evident mainly in variants of intermediate age, and not in recent or very old variants. The data fit a model in which there was a pulse of mutation between about 2,000 and 15,000 years ago in the ancestors of present-day Europeans and South Asians, during which these mutations were 30–40% more likely.
One possible explanation is that this is the legacy of a mutator allele (that is, a mutation that increases the rate of de novo mutation in some or all categories) that appeared and survived in the population for several thousand years before going extinct (Figure 1). It is not known how frequently such alleles arise, or to what extent they drive differences in the mutation spectrum: however it does seem that under certain conditions they can survive for a long time and have lasting effects (Seoighe and Scally, 2017). In addition to a mutator allele that would have increased the relative rate of the TCC → TTC mutation, it is possible that other mutator alleles that had smaller effects and/or survived for shorter times in different populations may be responsible for less prominent differences in the mutation spectrum.
Figure 1
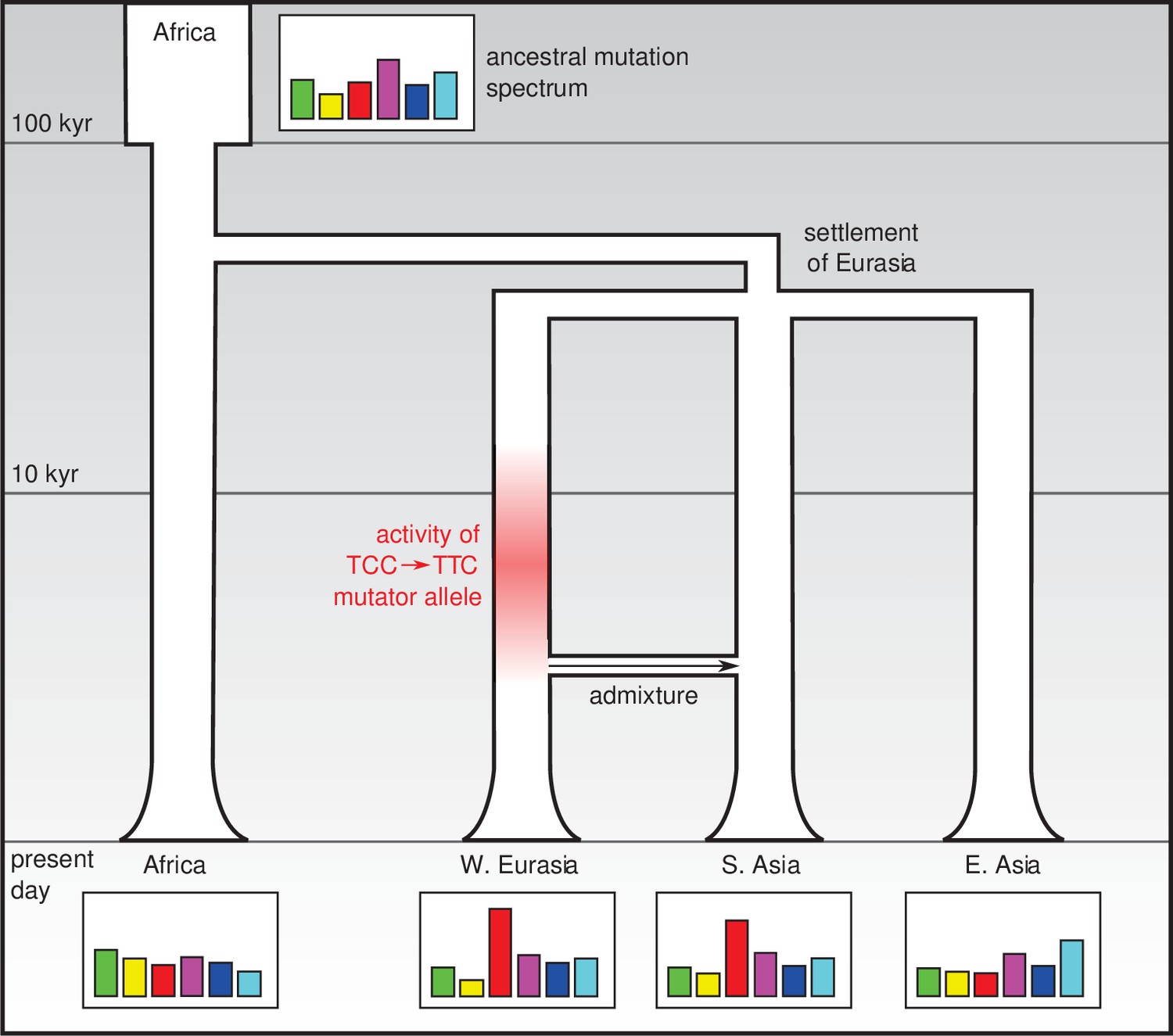
Changes in the genetic mutation spectrum in humans.
Modern humans originated in Africa (top left) and spread into Eurasia in a series of migrations 50,000–80,000 years ago. Subsequent migrations within Eurasia (such as the migration between West Eurasia and South Asia around 3000 years ago that is shown here; Reich et al., 2009) led to secondary genetic contact and admixture. The mutation spectrum (represented abstractly here) evolved separately in each population after divergence, perhaps due to the effect of mutator alleles: Harris and Pritchard propose that such a mutator allele could have been responsible for an increase in TCC → TTC mutations between 2,000 and 15,000 years ago (red shading) which influenced the mutation spectra of present-day Europeans and South Asians.
These findings are also relevant to the question of whether or not the overall rate of genomic mutation varies between populations and over time. There is evidence that the mean rate has probably not changed in at least the last 50,000 years (Fu et al., 2014), but we also know that the genome-wide mutation rate has slowed down when measured over timescales of millions of years (Moorjani et al., 2016). The results of Harris and Pritchard involve the relative rates of mutation in different sequence contexts, and so strictly speaking they do not tell us about variation in the overall rate, but they are suggestive of differences perhaps on the order of a few percent.
The next few years will see a substantial increase in the amount of available de novo mutation data, particularly from large-scale sequencing projects aimed at understanding the causes of rare genetic disease. These data will enable direct exploration of the factors determining mutation and its evolution within human populations, and shed further light on the questions addressed and raised by Harris and Pritchard.
References
-
De novo mutations in human genetic diseaseNature Reviews Genetics 13:565–575.https://doi.org/10.1038/nrg3241
Article and author information
Author details
Publication history
- Version of Record published: May 17, 2017 (version 1)
Copyright
© 2017, Scally
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,152
- views
-
- 257
- downloads
-
- 2
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Population Genetics: Global clues to the nature of genomic mutations in humans
eLife 6:e27605.
https://doi.org/10.7554/eLife.27605
Further reading
-
- Developmental Biology
- Evolutionary Biology
Despite rapid evolution across eutherian mammals, the X-linked MIR-506 family miRNAs are located in a region flanked by two highly conserved protein-coding genes (SLITRK2 and FMR1) on the X chromosome. Intriguingly, these miRNAs are predominantly expressed in the testis, suggesting a potential role in spermatogenesis and male fertility. Here, we report that the X-linked MIR-506 family miRNAs were derived from the MER91C DNA transposons. Selective inactivation of individual miRNAs or clusters caused no discernible defects, but simultaneous ablation of five clusters containing 19 members of the MIR-506 family led to reduced male fertility in mice. Despite normal sperm counts, motility, and morphology, the KO sperm were less competitive than wild-type sperm when subjected to a polyandrous mating scheme. Transcriptomic and bioinformatic analyses revealed that these X-linked MIR-506 family miRNAs, in addition to targeting a set of conserved genes, have more targets that are critical for spermatogenesis and embryonic development during evolution. Our data suggest that the MIR-506 family miRNAs function to enhance sperm competitiveness and reproductive fitness of the male by finetuning gene expression during spermatogenesis.
-
- Evolutionary Biology
- Immunology and Inflammation
CD4+ T cell activation is driven by five-module receptor complexes. The T cell receptor (TCR) is the receptor module that binds composite surfaces of peptide antigens embedded within MHCII molecules (pMHCII). It associates with three signaling modules (CD3γε, CD3δε, and CD3ζζ) to form TCR-CD3 complexes. CD4 is the coreceptor module. It reciprocally associates with TCR-CD3-pMHCII assemblies on the outside of a CD4+ T cells and with the Src kinase, LCK, on the inside. Previously, we reported that the CD4 transmembrane GGXXG and cytoplasmic juxtamembrane (C/F)CV+C motifs found in eutherian (placental mammal) CD4 have constituent residues that evolved under purifying selection (Lee et al., 2022). Expressing mutants of these motifs together in T cell hybridomas increased CD4-LCK association but reduced CD3ζ, ZAP70, and PLCγ1 phosphorylation levels, as well as IL-2 production, in response to agonist pMHCII. Because these mutants preferentially localized CD4-LCK pairs to non-raft membrane fractions, one explanation for our results was that they impaired proximal signaling by sequestering LCK away from TCR-CD3. An alternative hypothesis is that the mutations directly impacted signaling because the motifs normally play an LCK-independent role in signaling. The goal of this study was to discriminate between these possibilities. Using T cell hybridomas, our results indicate that: intracellular CD4-LCK interactions are not necessary for pMHCII-specific signal initiation; the GGXXG and (C/F)CV+C motifs are key determinants of CD4-mediated pMHCII-specific signal amplification; the GGXXG and (C/F)CV+C motifs exert their functions independently of direct CD4-LCK association. These data provide a mechanistic explanation for why residues within these motifs are under purifying selection in jawed vertebrates. The results are also important to consider for biomimetic engineering of synthetic receptors.




{kind=link}