Mechanism of chiral proofreading during translation of the genetic code
- Centre for Cellular and Molecular Biology, Council for Scientific and Industrial Research, India
Figures
Figure 1 with 2 supplements
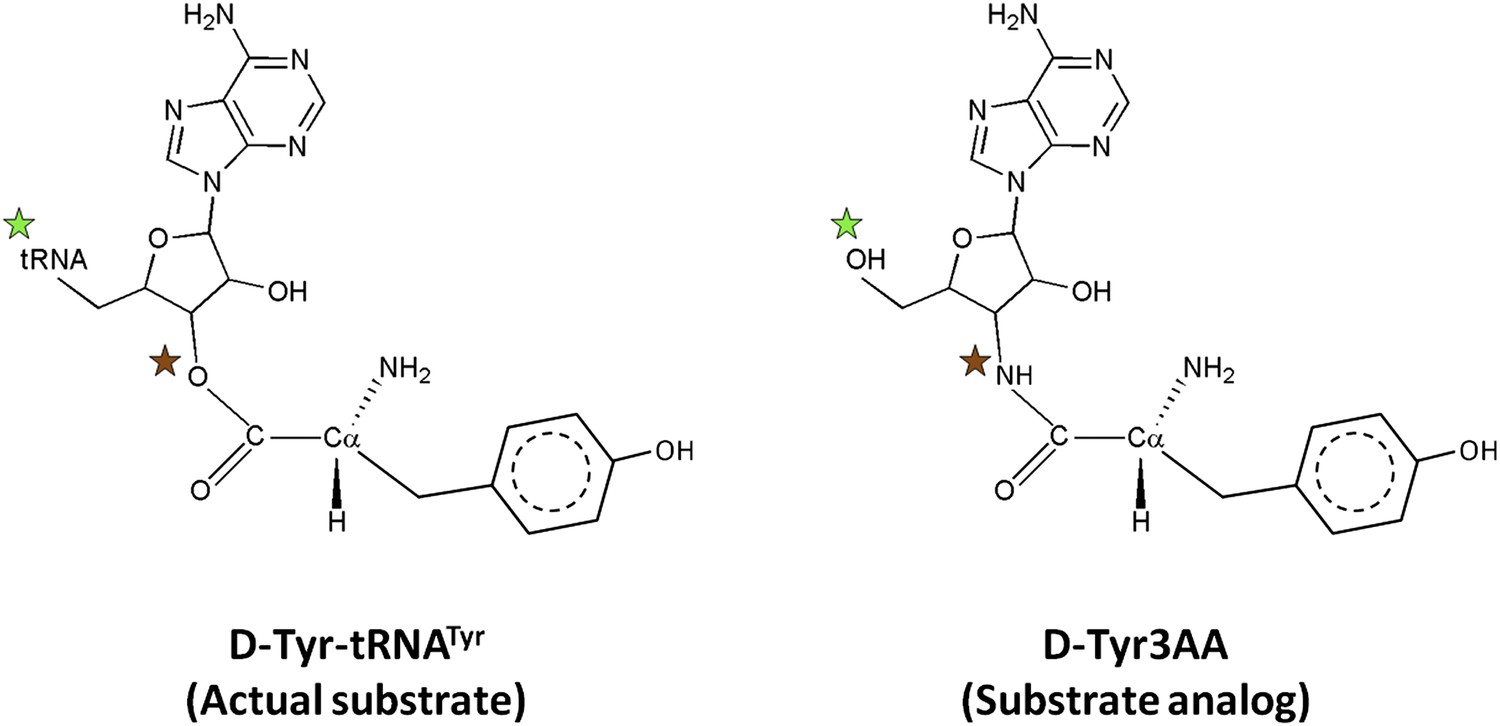
Comparison of the actual substrate with the analog used in this study.
( ) The 5′-OH is linked to tRNA in the actual substrate, whereas it is free in D-Tyr3AA. (
) The 5′-OH is linked to tRNA in the actual substrate, whereas it is free in D-Tyr3AA. ( ) The ester bond in the real substrate is replaced by an amide bond in the analog D-Tyr3AA to make it non-hydrolyzable.
) The ester bond in the real substrate is replaced by an amide bond in the analog D-Tyr3AA to make it non-hydrolyzable.
Figure 1—figure supplement 1
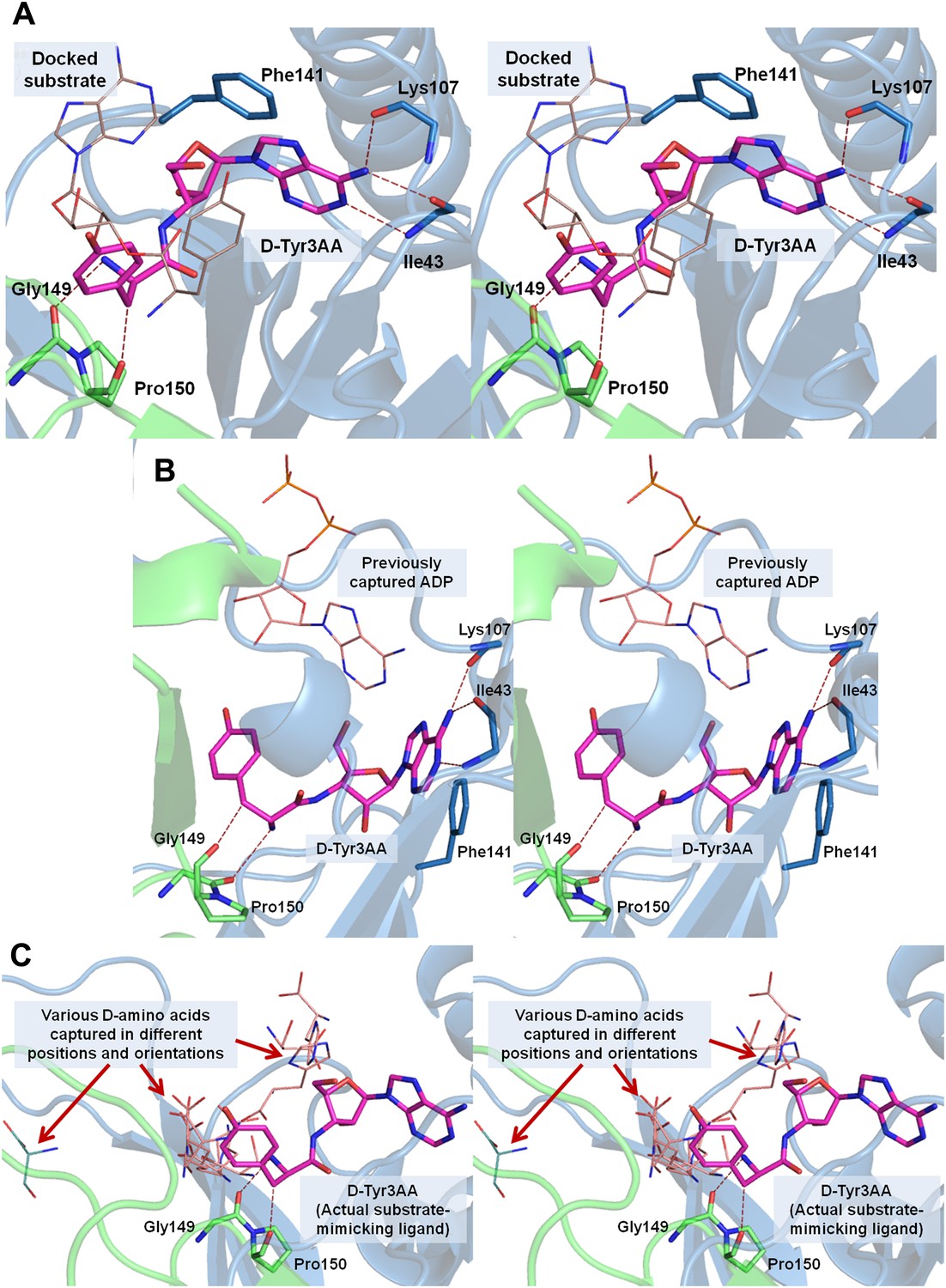
Stereoscopic images showing the positions of earlier modeled ligands with respect to the cognate substrate-mimicking analog D-Tyr3AA in DTD.
(A) Comparison of the docked substrate (Lim et al., 2003) with D-Tyr3AA captured in the co-crystal structure. In their study, Lim et al. have docked the entire substrate that is D-Tyr-tRNA onto Haemophilus influenzae DTD but here only the terminal adenosine is shown for the sake of comparison. To prepare the figure, the ligand has been placed based on the stereoscopic image provided by Lim et al. (2003). The docked substrate complex does not match with the D-Tyr3AA as found in the experimental structure presented here. (B) The position of ADP (Bhatt et al., 2010) is actually completely outside the pocket when compared to D-Tyr3AA complex. (C) The positions and orientations of various D-amino acids captured (Bhatt et al., 2010) with respect to the cognate substrate analog D-Tyr3AA. None of the D-amino acids is found to be located in the position where the chiral discrimination occurs (the distance between Cα of the D-amino acids and Cα of D-Tyr3AA ranges from 3.39 Å to 13.94 Å). The site of binding as well as the orientation of D-amino acids with respect to the enzyme is highly variable. In some cases, the carboxylate group points towards the enzyme, whereas in some cases it projects outward. Considering that in the actual substrate, the D-amino acid would be linked to the tRNA, a multiple binding mode is highly improbable as the orientation and position of the D-amino acid would be fixed by the binding of terminal adenosine.
Figure 1—figure supplement 2
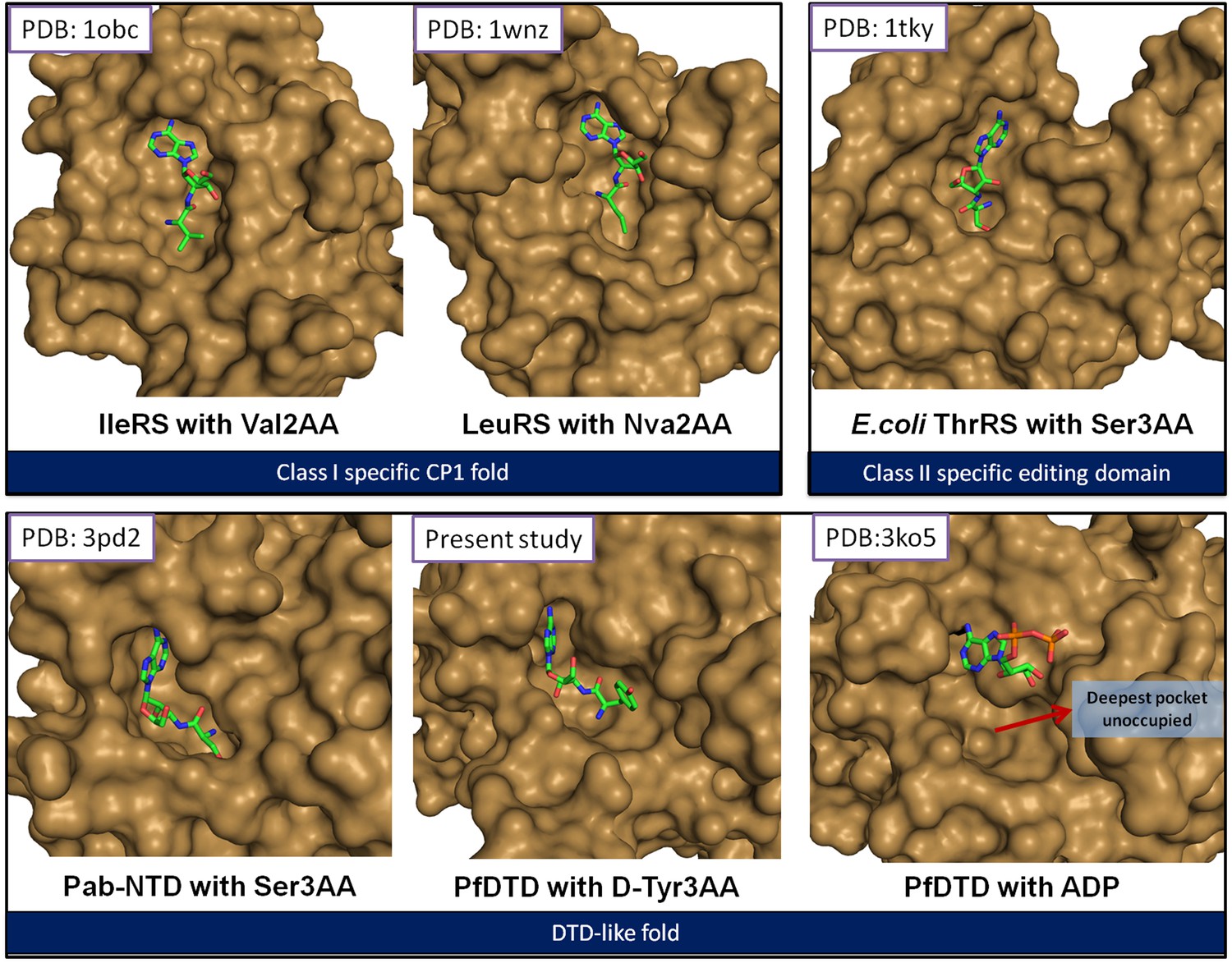
The reported structures of proofreading domains with substrate-mimicking analogs.
The substrate analog-bound structures that represent the biologically relevant complexes of proofreading domains invariably occupy the deepest available pocket having a striking surface complementarity. This can be clearly seen in the well-studied cases of Class I-specific CP1 domains, Class II-specific editing domain of E. coli ThrRS and Pab-NTD. The D-Tyr3AA complex presented in this study also shows these features evidently. However, the ADP complex (Bhatt et al., 2010) shows ADP clinging onto the surface, leaving the deepest pocket unoccupied. It may also be noted here that ADP is not a substrate for this enzyme.
Figure 2 with 6 supplements

Structure of DTD in complex with D-Tyr3AA.
(A) A (2Fo–Fc) map contoured at 1.2σ clearly showing unambiguous density for the ligand D-Tyr3AA from crystal form I solved at 1.86 Å resolution. (B) Dimeric DTD with the two monomers shown in green and cyan. The conserved–SQFTL–and–NXGP(V/F)T–motifs are depicted in violet and orange respectively. The ligand binds in the two active sites located at the dimer interface. The two tRNAs have been schematically represented. (C) Surface representation showing D-Tyr3AA in the pocket. Inset is a magnified image showing the side chain of D-tyrosine protruding out of the pocket. The ligand has been colored according to the B-factors. (D) Structural rearrangements in the substrate pocket upon D-Tyr3AA binding highlighting the plasticity of the active site. The apo is shown in green and the complex is shown in purple. The ligand has been made transparent for clarity. (E) Stereoscopic representation showing the interactions between the ligand and the active site residues (* indicates residues from the other monomer).
Figure 2—figure supplement 1
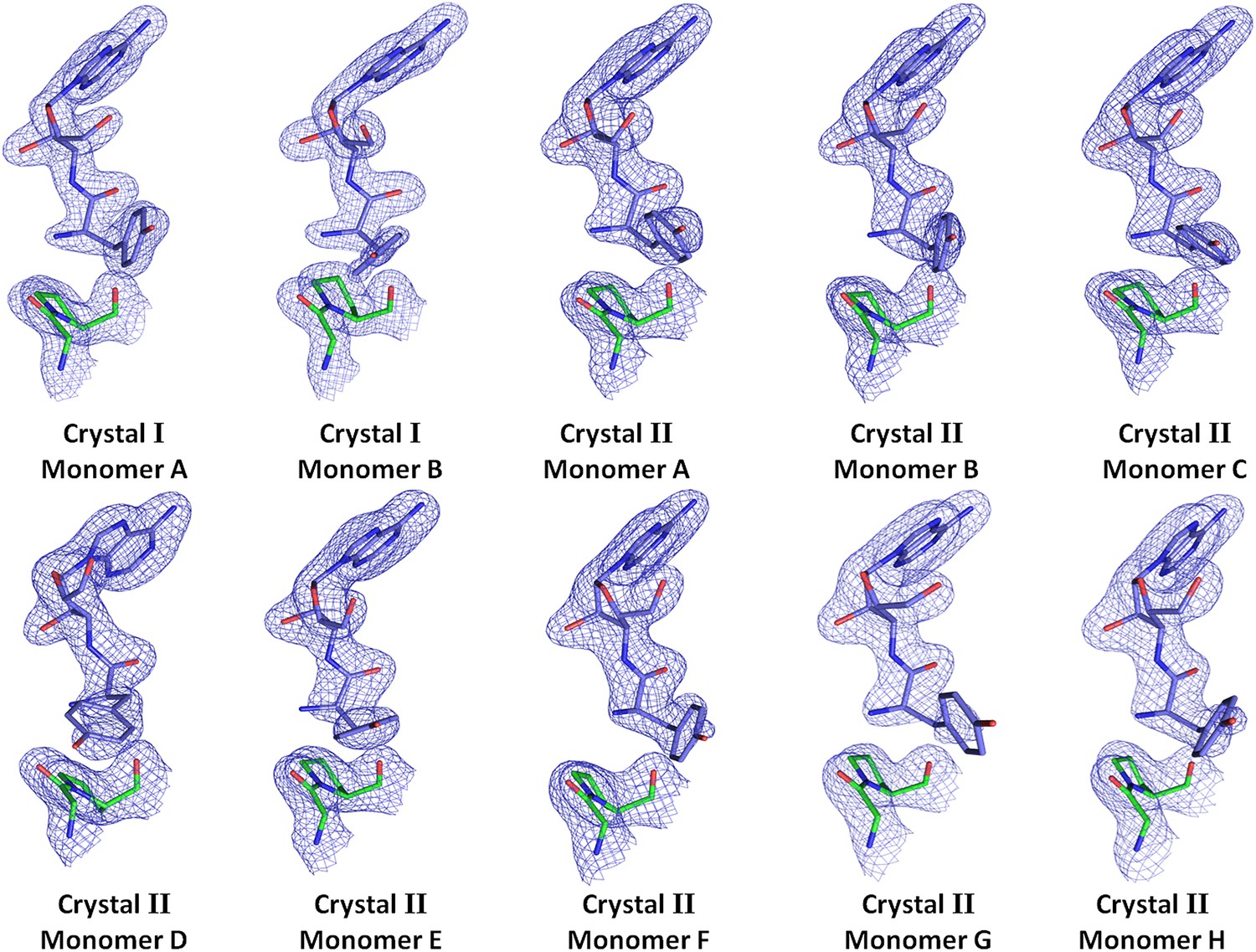
Electron density for the ligand in all observations.
(2Fo–Fc) maps contoured at 1.2σ for all monomers from crystal forms I and II showing clear unambiguous densities for the ligand D-Tyr3AA.
Figure 2—figure supplement 2
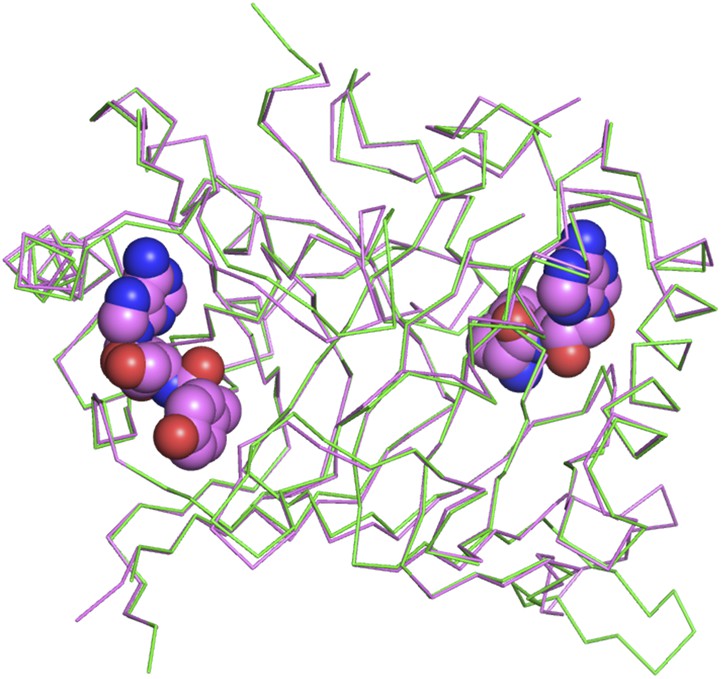
Superimposition of D-Tyr3AA-bound complex structure of PfDTD (pink) on the apo structure (green).
The complex structure overlaps with the apo structure with an r.m.s.d. of 0.41 Å over 260 Cα atoms.
Figure 2—figure supplement 3
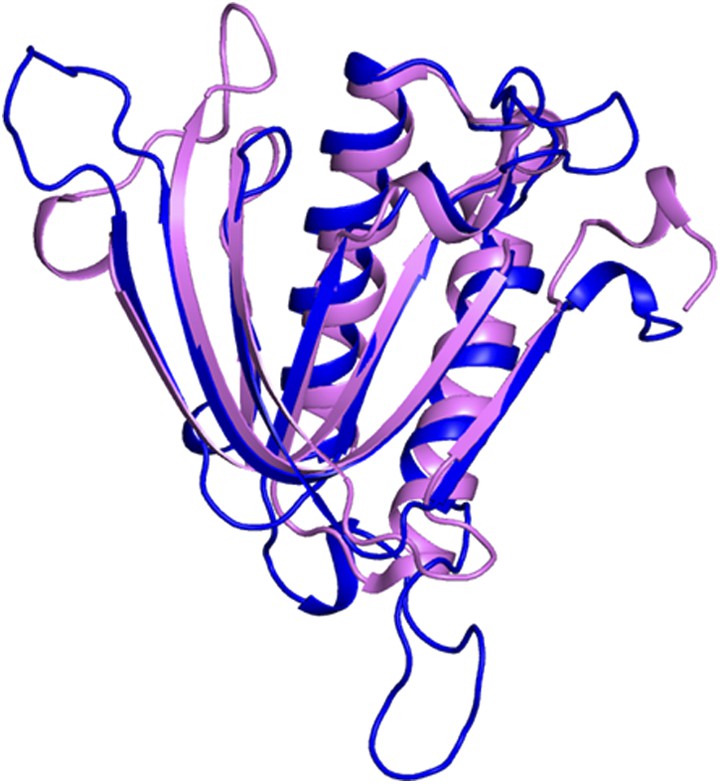
Superimposition of PfDTD on Pab-NTD.
PfDTD has been shown in blue and Pab-NTD is depicted in pink. The two structures overlap with an r.m.s.d. of 1.65 Å over 118 Cα atoms.
Figure 2—figure supplement 4
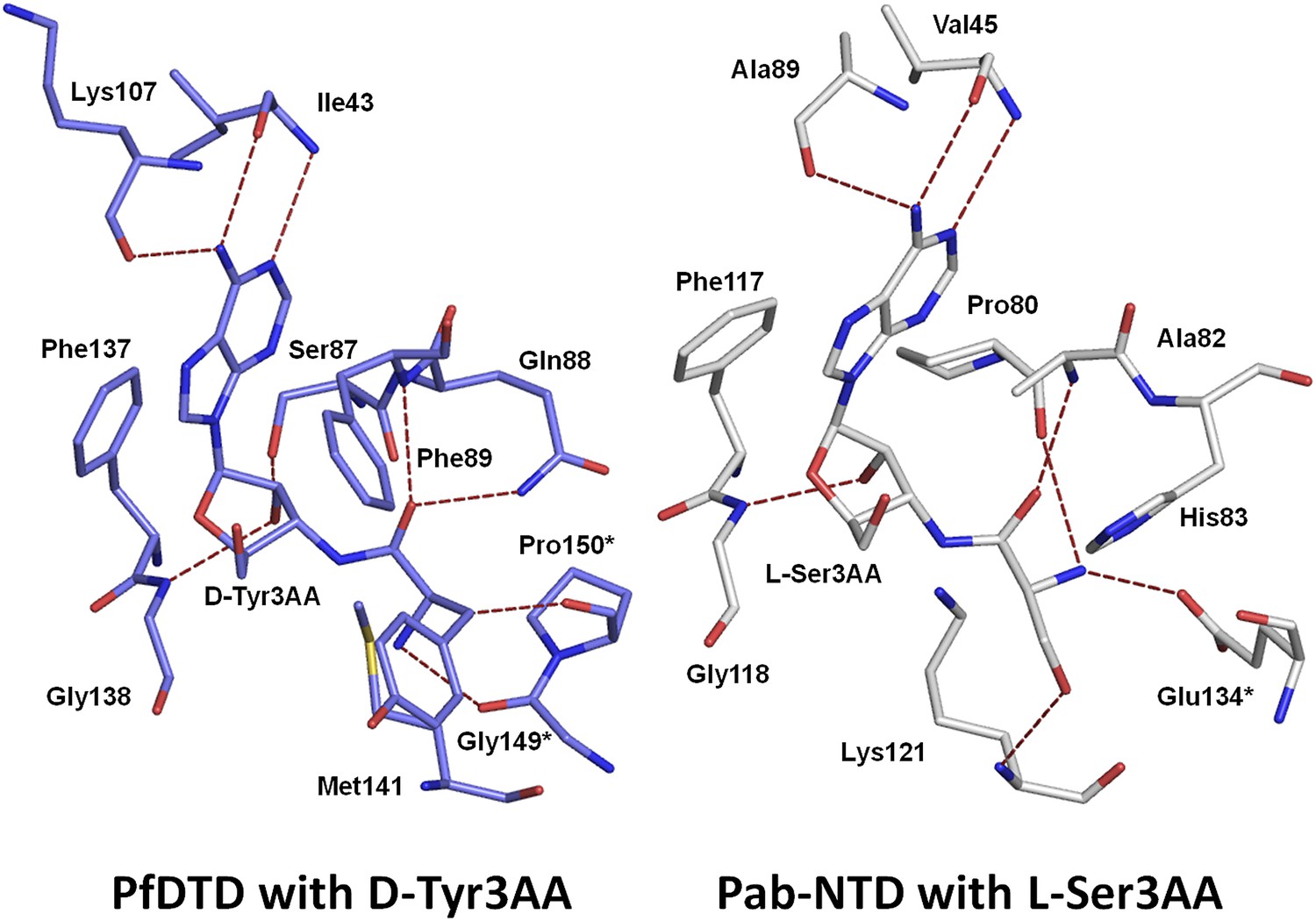
Comparison of ligand interaction in PfDTD and Pab-NTD.
The adenine is recognized by a conserved set of interactions in both PfDTD and Pab-NTD, including an invariant Phe residue which stacks with the adenine base.
Figure 2—figure supplement 5
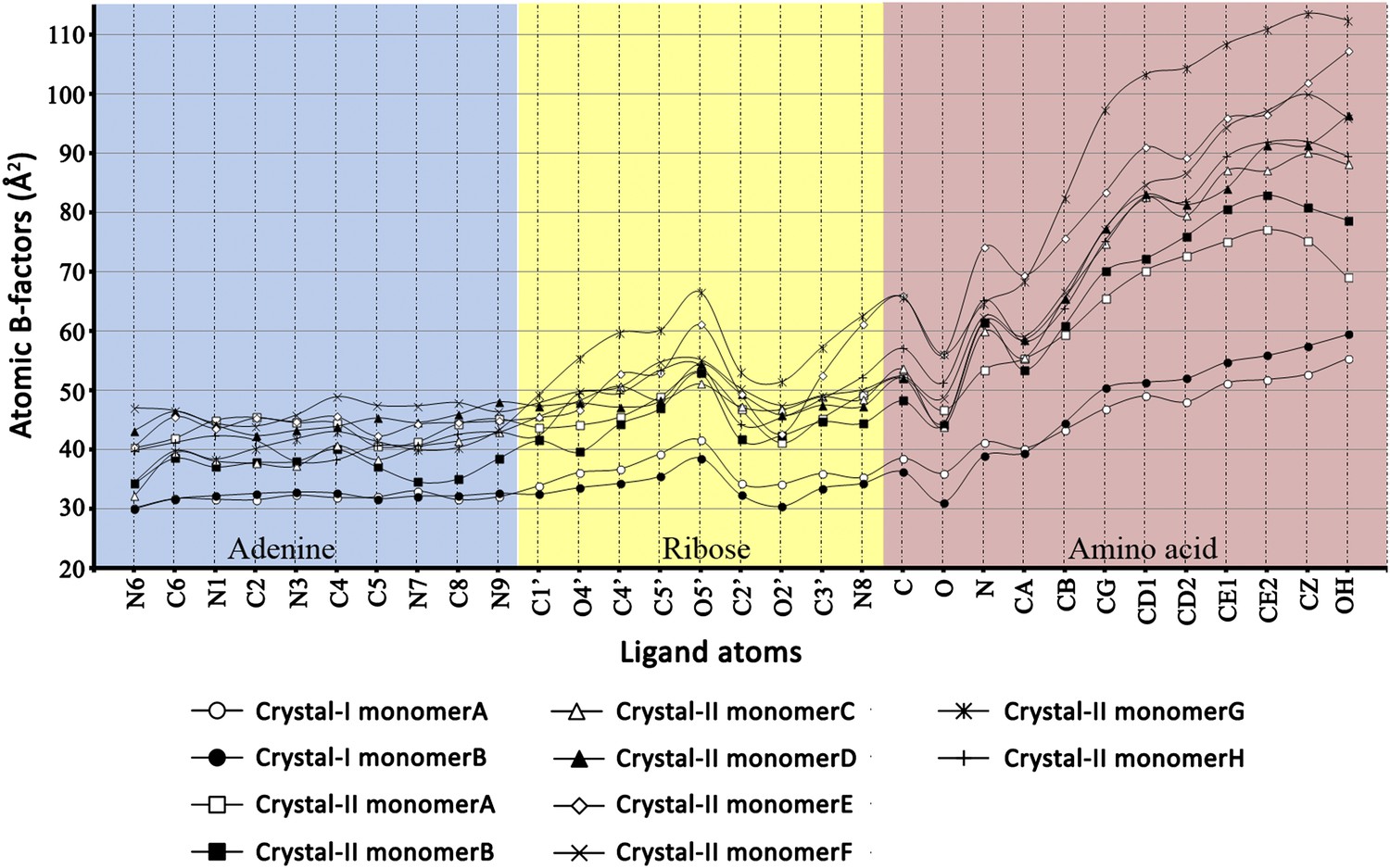
Atomic B-factors plotted for all the ligand atoms from both crystal forms I and II.
The atomic B-factor is given by the equation: Bi = 8π2Ui2 (where Ui is the mean square displacement of atom i). It is a measure of atomic displacement. High B-factors indicate flexibility while ordered regions have low B-factors. A sharp rise in the B-factors can be observed in the amino acid moiety beyond the Cβ atom. Another peak is observed around the C5′ and 5′-OH of ribose since it is placed out of the pocket. A dip at the carbonyl oxygen of the amino acid highlights the strong recognition of this atom in the active site pocket.
Figure 2—figure supplement 6
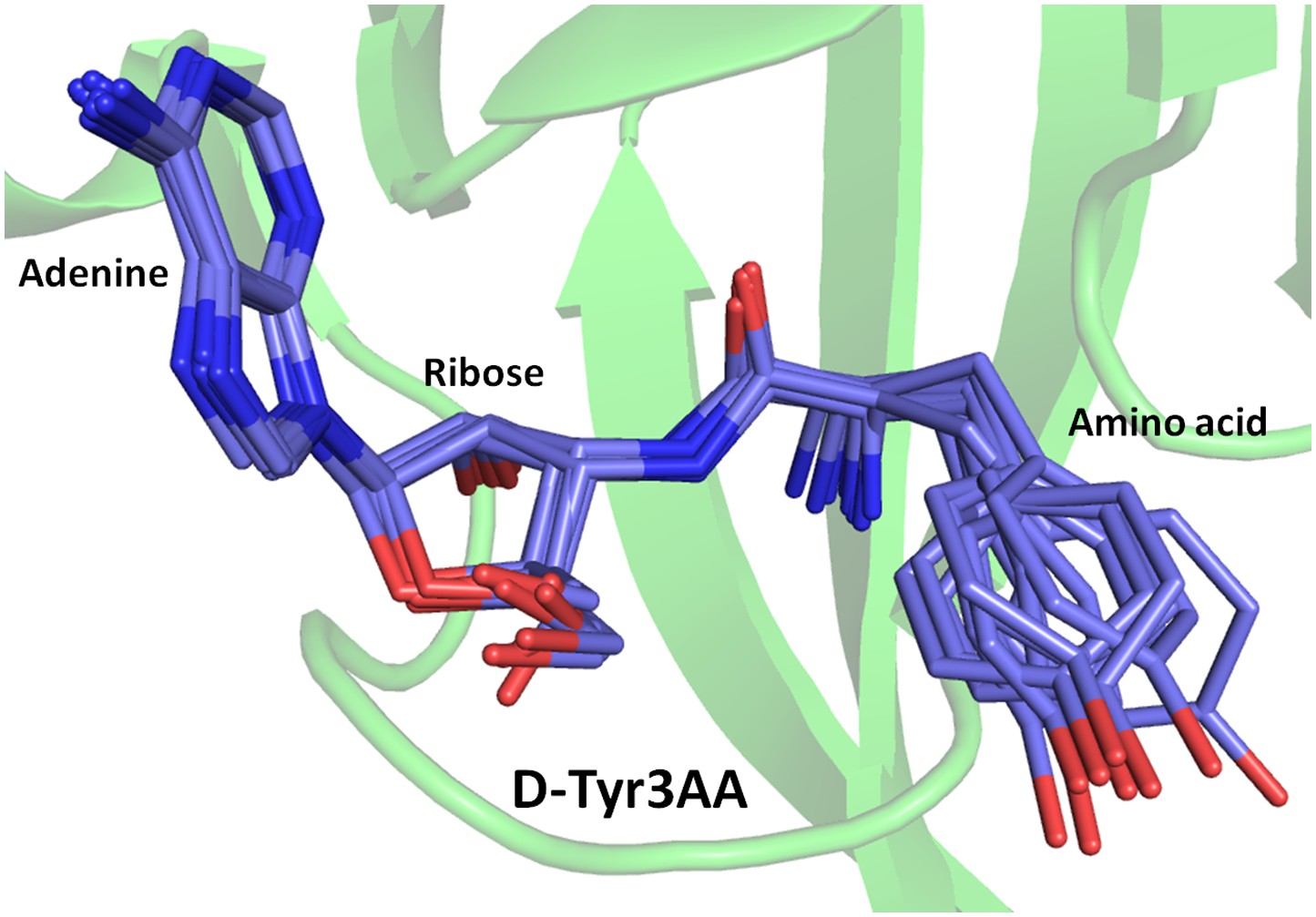
Superimposition of D-Tyr3AA from all monomers of crystal forms I and II.
The adenine and ribose superimpose on top of each other very well. However, the amino acid moiety shows significant deviations. The maximum variation is observed beyond the Cβ atom of the substrate.
Figure 3 with 2 supplements
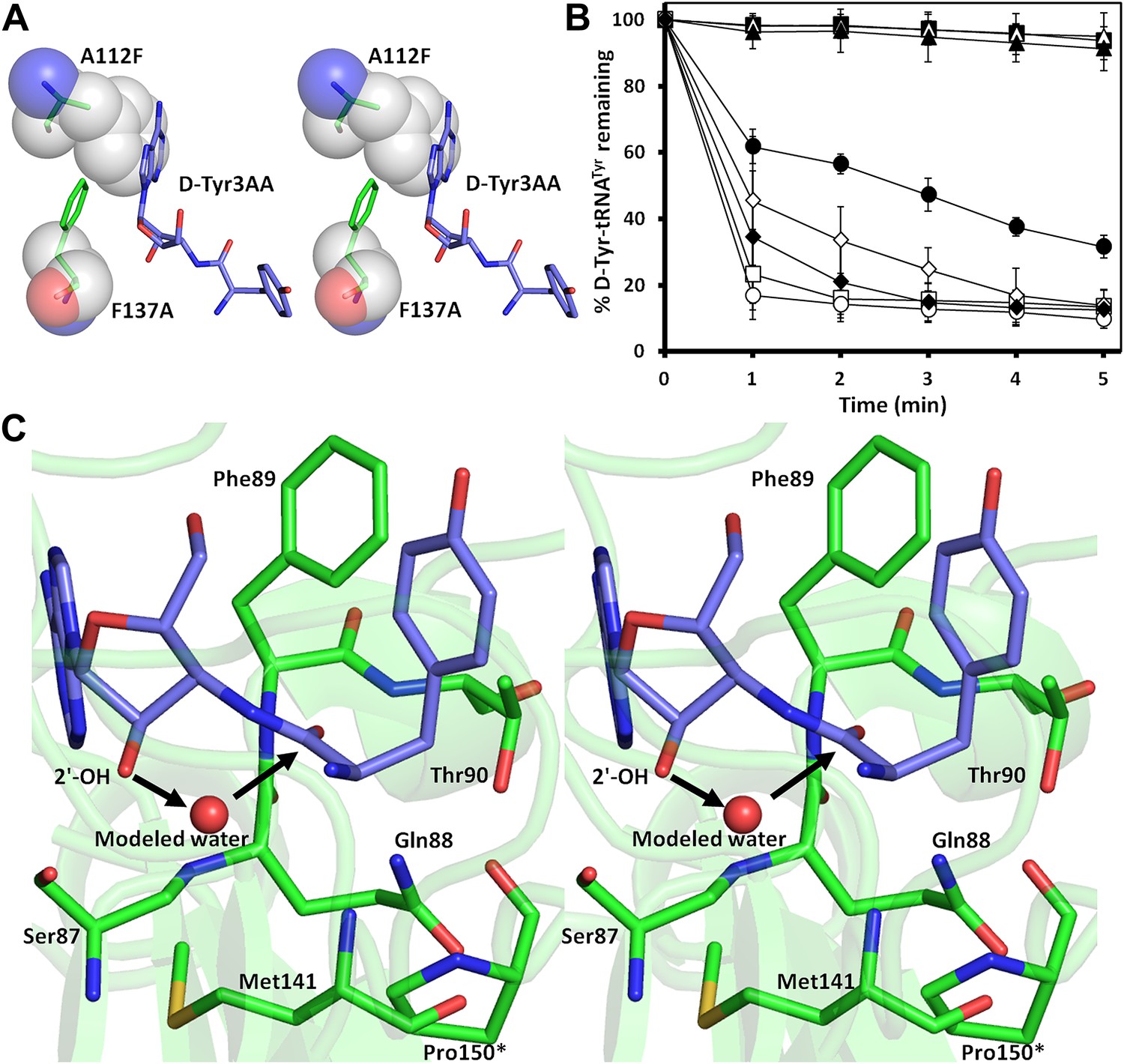
Mutational analysis of the active site residues.
(A) Stereoscopic depiction showing mutations generated in the adenine-binding pocket: Stick representation is used for wild-type residues while mutants are depicted in spheres. Phe137 was mutated to Ala and Ala112 was mutated to Phe. (B) Deacylation of D-Tyr-tRNATyr by buffer ( ), wild-type PfDTD (
), wild-type PfDTD ( ), F137A (
), F137A ( ), A112F (
), A112F ( ), S87A (
), S87A ( ), S87P (
), S87P ( ), Q88A (
), Q88A ( ) and T90A (
) and T90A ( ). 500 pM enzyme concentration was used for the assays. (C) Stereoscopic image showing all the protein side chains within 6 Å of the susceptible bond of the substrate. A water molecule has been modeled based on Pab-NTD complex structure. The water is positioned at a distance of 2.61 Å from the 2′-OH and 2.79 Å from the scissile bond of D-Tyr3AA. In the absence of any protein side chain playing a role in catalysis, a substrate-assisted mechanism is proposed involving the role of 2′-OH of tRNA in activating a water molecule as suggested in case of Pab-NTD.
). 500 pM enzyme concentration was used for the assays. (C) Stereoscopic image showing all the protein side chains within 6 Å of the susceptible bond of the substrate. A water molecule has been modeled based on Pab-NTD complex structure. The water is positioned at a distance of 2.61 Å from the 2′-OH and 2.79 Å from the scissile bond of D-Tyr3AA. In the absence of any protein side chain playing a role in catalysis, a substrate-assisted mechanism is proposed involving the role of 2′-OH of tRNA in activating a water molecule as suggested in case of Pab-NTD.
Figure 3—figure supplement 1
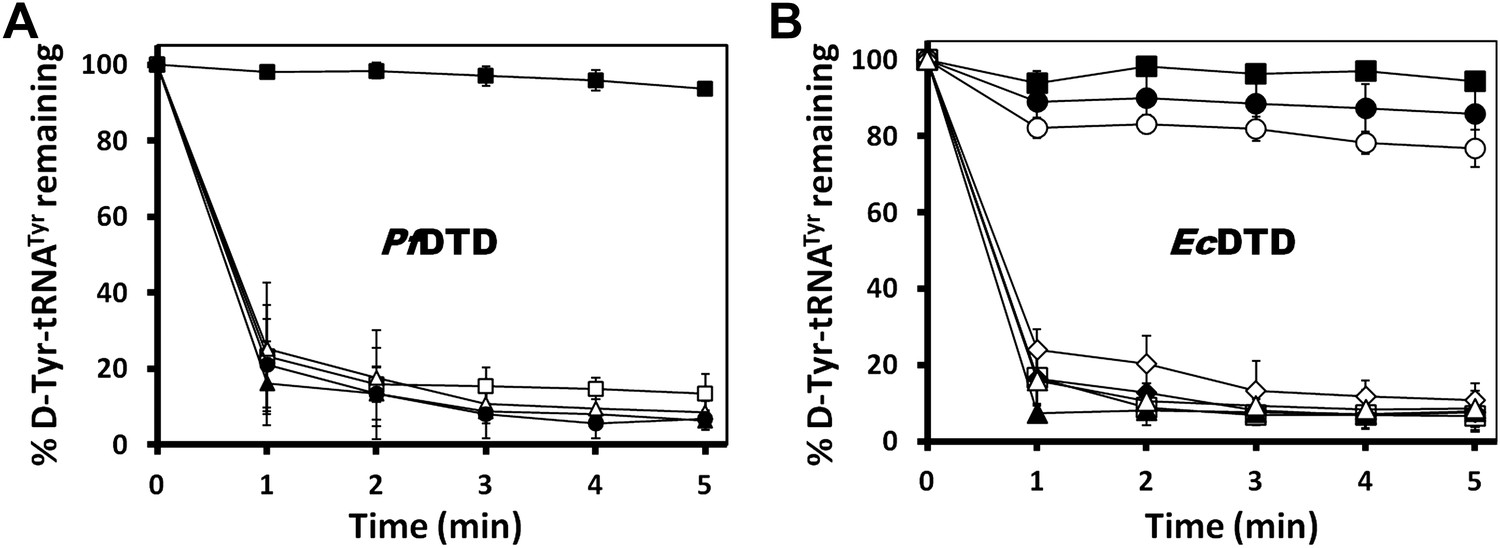
Mutational analysis of the active site residues in PfDTD and EcDTD.
(A) Deacylation of D-Tyr-tRNATyr by buffer (), wild type PfDTD (), Q88E (), Q88N (), T90S (). 500 pM enzyme concentration was used for all assays. (B) Deacylation of D-Tyr-tRNATyr by buffer (), wild-type EcDTD (), F125A (), A102F (), S77A (), S77P (), Q78A (), T80A (). 50 nM enzyme concentration was used for all assays.
Figure 3—figure supplement 2
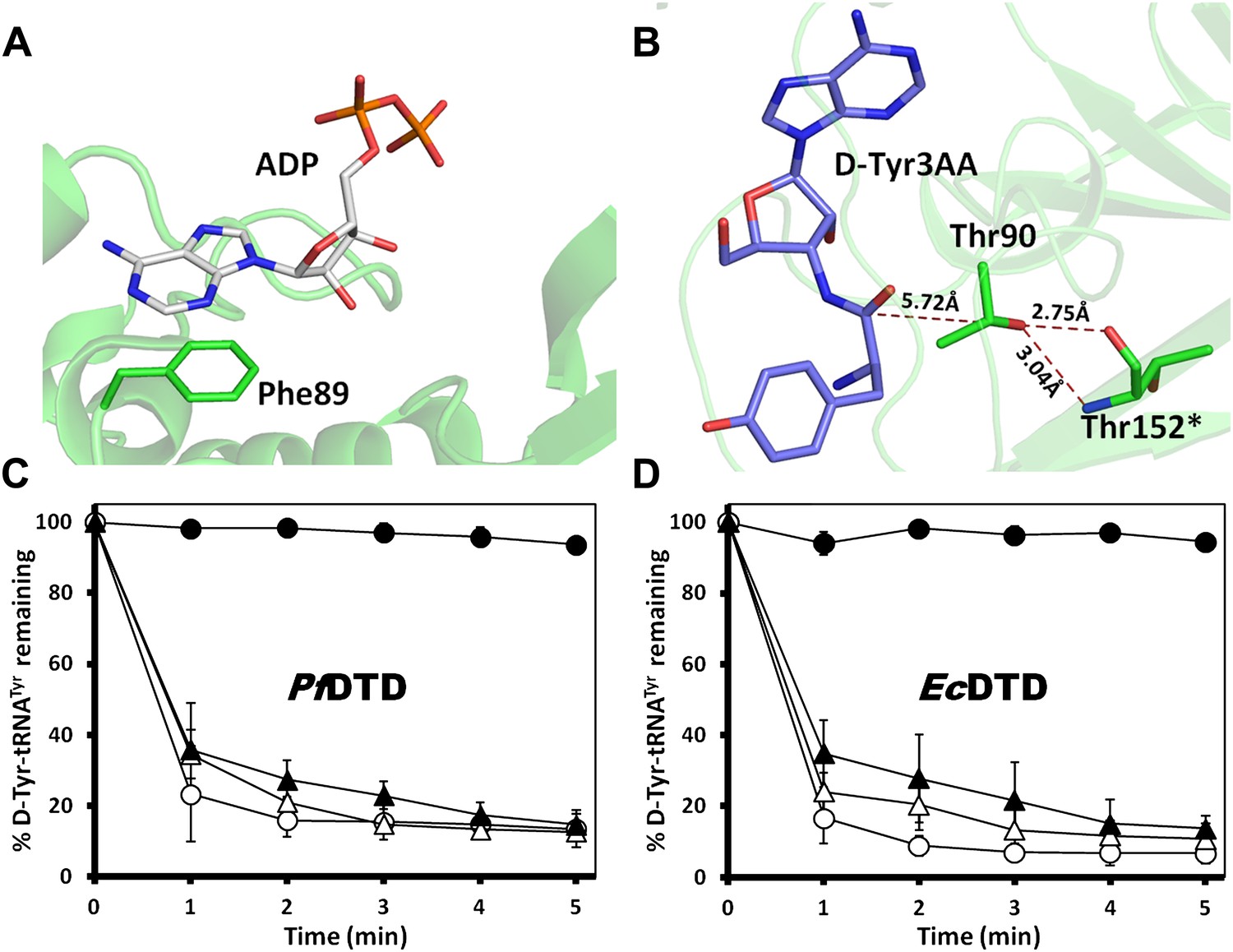
Mutational data on the earlier identified binding modes in DTD.
(A) The ADP-bound structure as reported earlier shows only one conserved interaction where the adenine base stacks with Phe89 while the phosphate tail hangs out (PDB id: 3KO5). (B) Thr90 that was earlier proposed to be the catalytic residue has its γ-hydroxyl group oriented away from the point of attack and is tightly held by highly conserved interactions with Thr152 main chain atoms from the other monomer. (C) Deacylation of D-Tyr-tRNATyr by buffer (), wild type PfDTD (), F89A () and T90A (). 500 pM of enzyme was used for each assay. Although T90A deacylation curve has been shown in Figure 3B, it is shown again here for immediate reference. (D) Deacylation of D-Tyr-tRNATyr by buffer (), wild type EcDTD (), F79A () and T80A (). 50 nM of enzyme was used for each assay. Although T80A deacylation curve has been shown in Figure 3—figure supplement 1B, it is shown again here for immediate reference.
Figure 4 with 1 supplement
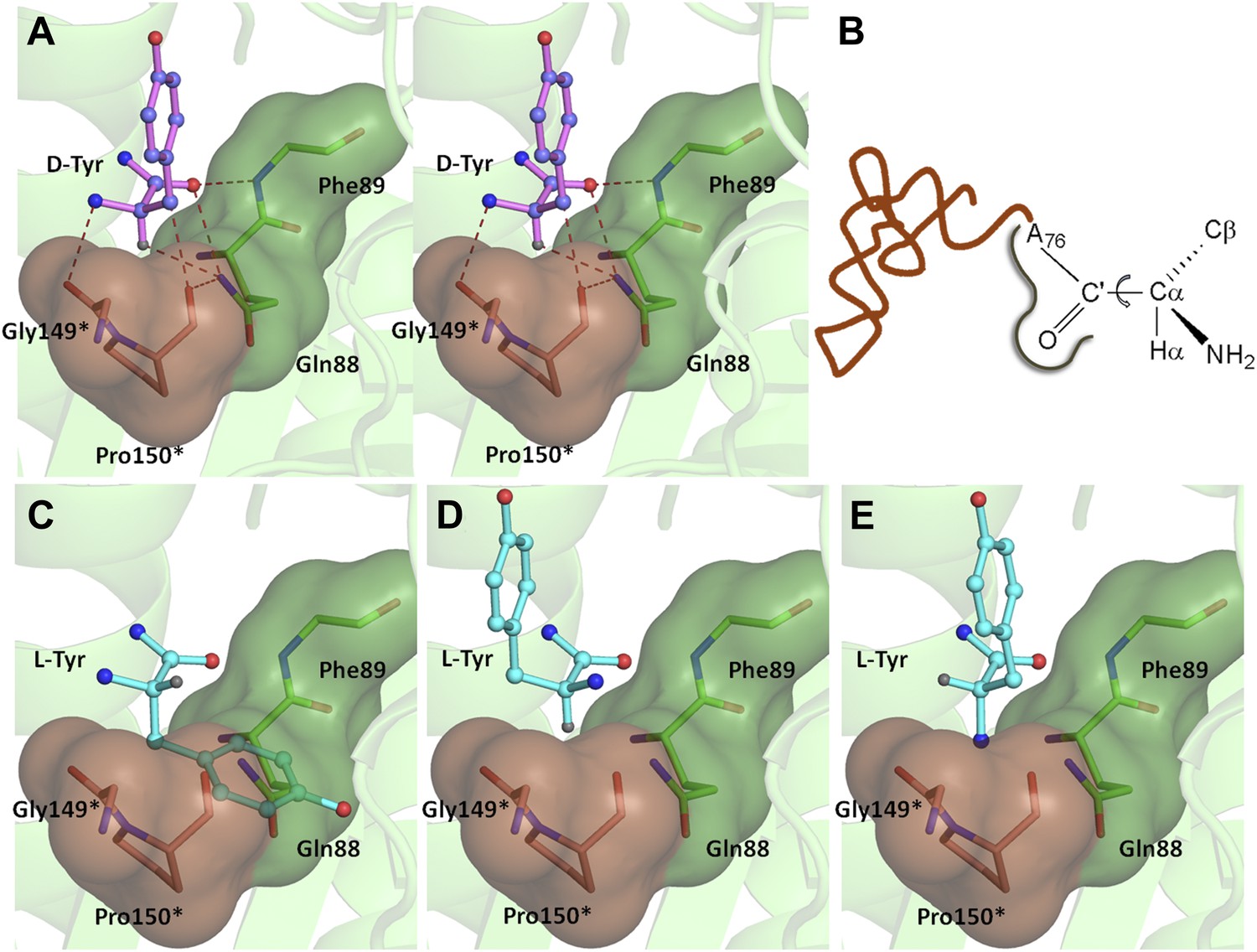
Mechanism of L-chirality rejection.
The cross-subunit Gly-cisPro motif is shown in brown. (A) Stereoscopic representation showing the conformation of D-amino acid observed in the pocket. (B) The adenosine moiety and the carbonyl oxygen are tightly fixed. The only allowed flexibility would be the torsion around Cα-C′ bond. This rotation gives rise to three theoretical possibilities of binding an L-amino acid. (C) Conformation I: the side chain swaps positions with Hα, severe short contacts of the side chain atoms including Cβ with active site residues can be seen. (D) Conformation II: the side chain swaps positions with NH2 group, short contact of side chain with C75 of tRNA, also Cβ is 2.56 Å from amide nitrogen (N8) of the substrate. (E) Conformation III: the NH2 group swaps positions with Hα, non-polar side chain of Pro150 provides unfavorable environment for NH2 group.
Figure 4—figure supplement 1
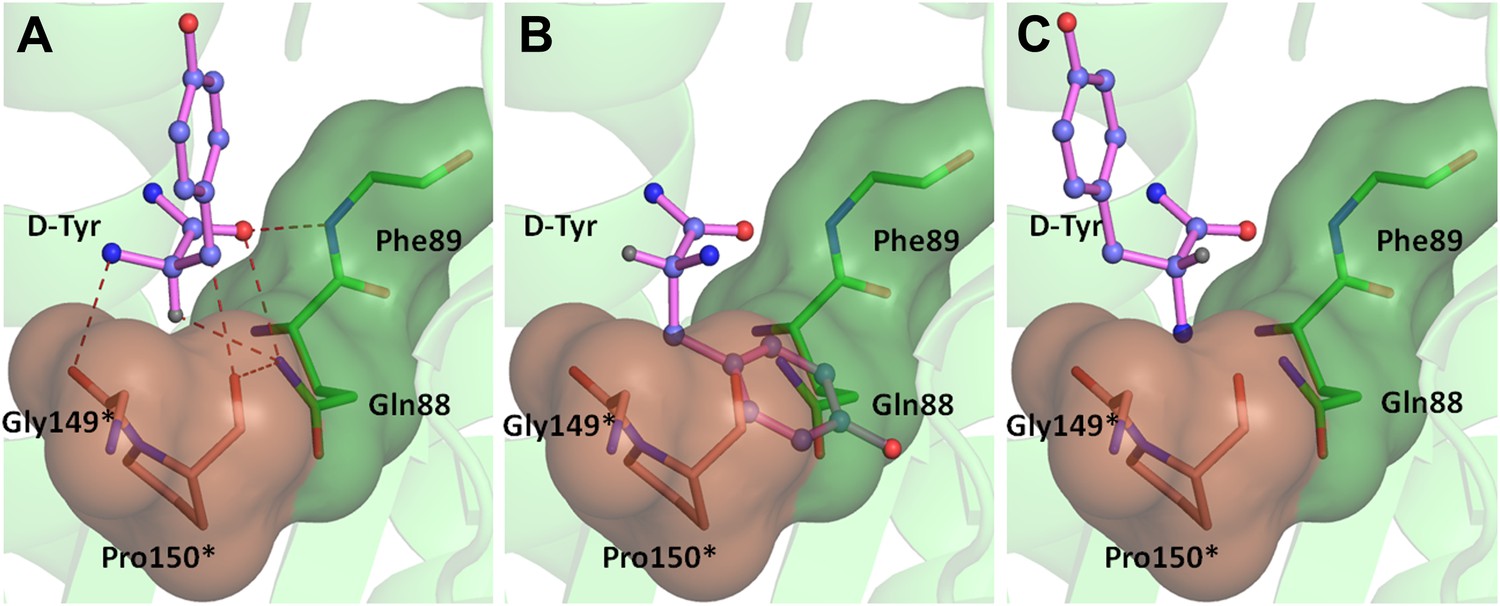
Theoretically possible modes of D-amino acid binding.
The cross-subunit Gly-cisPro motif is shown in brown. (A) The conformation of D-amino acid observed in the pocket; all the groups on the chiral Cα are captured. (B) Possibility 2: side chain occupies the position of Hα, severe short contacts of the side chain atoms including Cβ with active site residues can be seen. (C) Possibility 3: side chain occupies the position of NH2, the side chain would have short contacts with C75 of tRNA, Cβ would have short contacts with N8 and polar NH2 would be close to the non-polar Pro150 side chain.
Figure 5 with 3 supplements
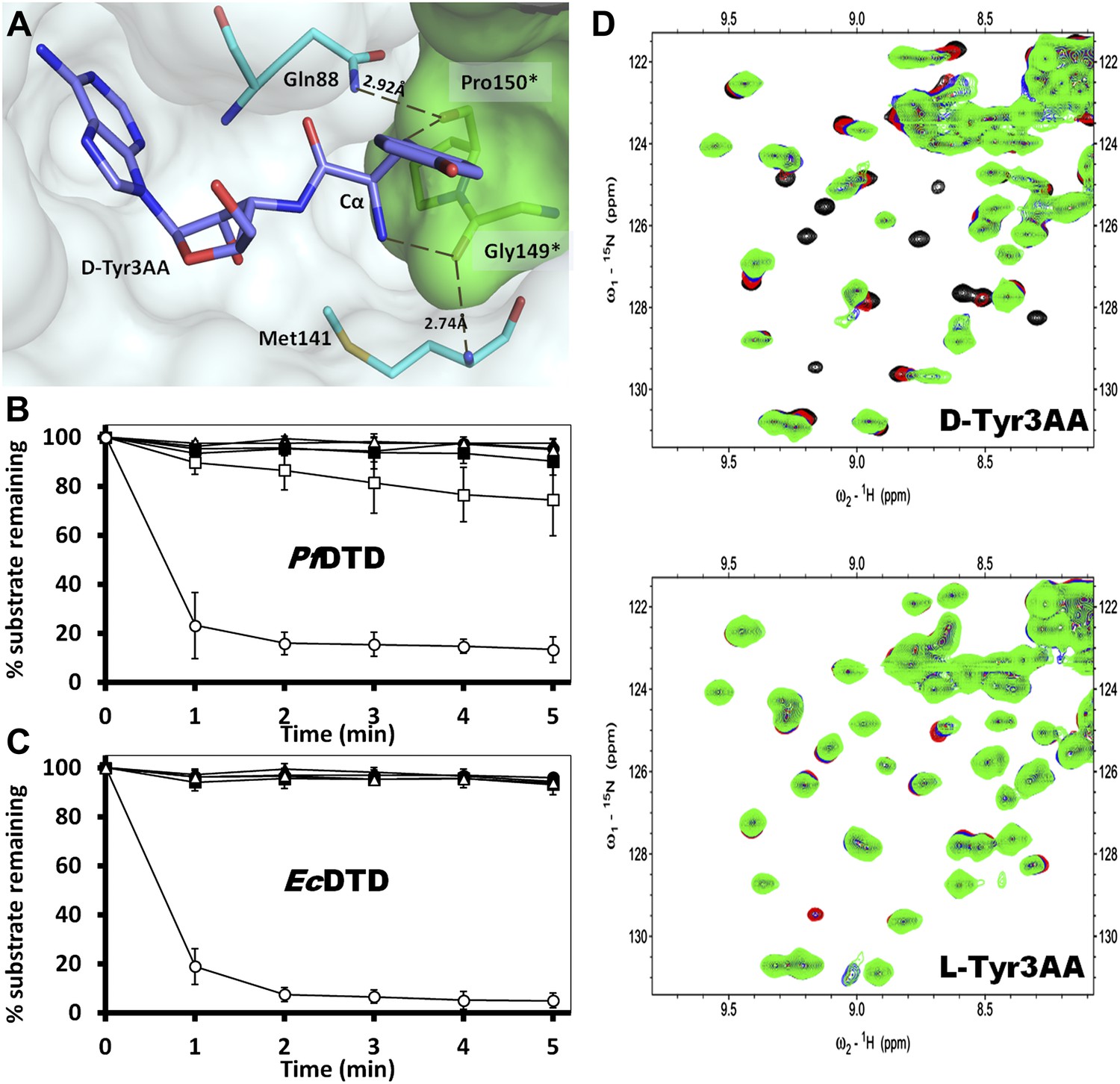
Strict configurational specificity of DTD.
(A) The Gly-cisPro motif from one monomer protrudes into the active site of the other monomer and cradles the chiral center of the substrate and provides basis for configuration selection. The carbonyl oxygens are tightly positioned by cross-subunit interactions. (B) Deacylation of L-Tyr-tRNATyr by buffer (), 500 pM (), 5 nM (), 50 nM (), 500 nM () PfDTD and D-Tyr-tRNATyr deacylation by 500 pM PfDTD (). (C) L-Tyr-tRNATyr deacylation by buffer (), 50 nM (), 500 nM (), 5 μM () EcDTD and D-Tyr-tRNATyr deacylation by 50 nM EcDTD (). (D) Excerpts of overlay of 2D 15N-1H TROSY obtained with 0.2 mM PfDTD (black) and upon addition of 1 mM (red), 2 mM (blue), 3 mM (green) D-Tyr3AA and L-Tyr3AA.
Figure 5—figure supplement 1
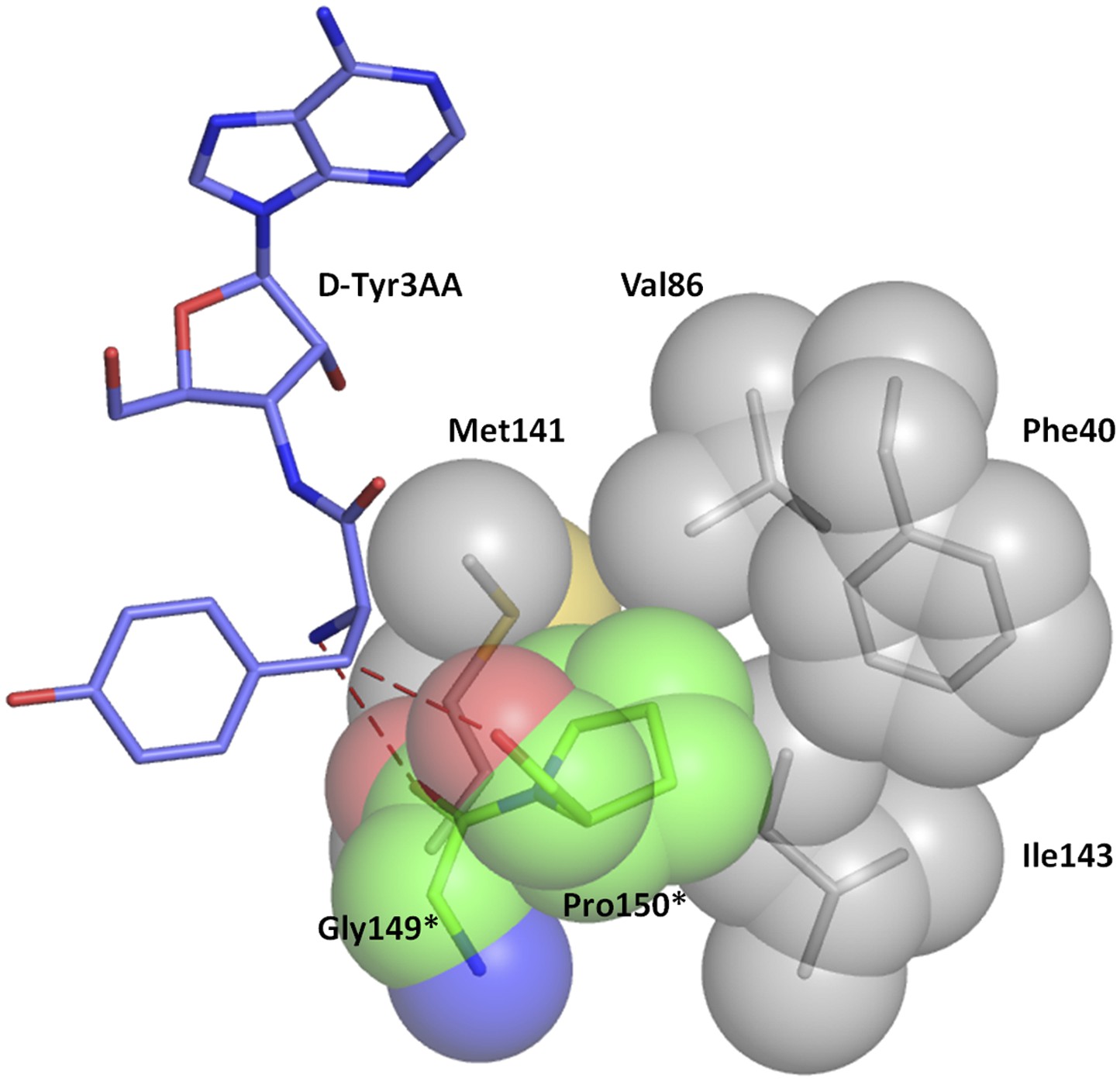
Hydrophobic base for Pro150 side chain.
The Pro150 side chain is supported by a hydrophobic base comprising of Phe40, Val86, Met141, and Ile143 side chains from the other monomer.
Figure 5—figure supplement 2
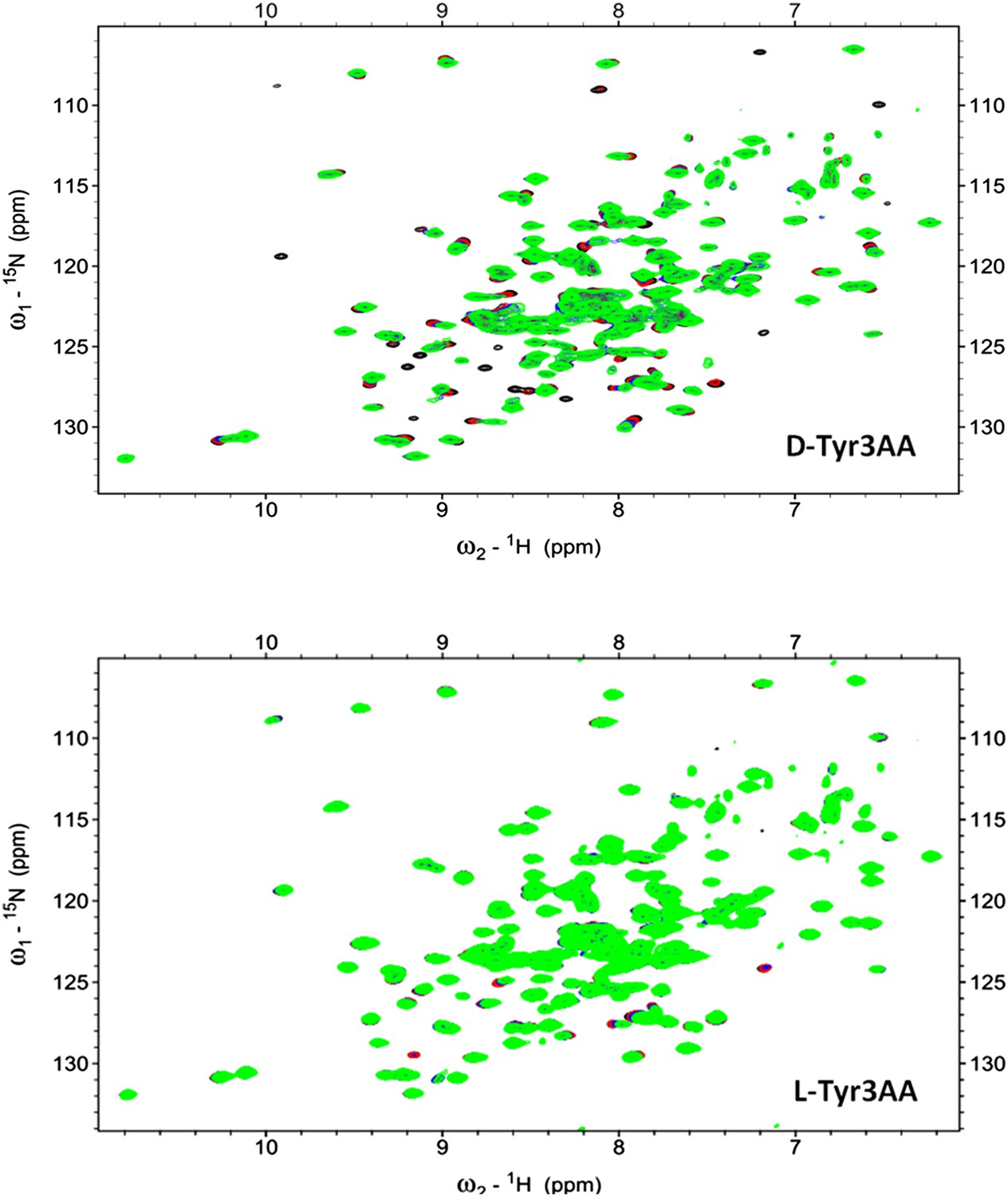
2D 15N-1H TROSY of PfDTD with D-Tyr3AA and L-Tyr3AA.
Overlay of 2D 15N-1H TROSY obtained with 0.2 mMPfDTD (black) and upon addition of 1 mM (red), 2 mM (blue), 3 mM (green) D-Tyr3AA and L-Tyr3AA.
Figure 5—figure supplement 3
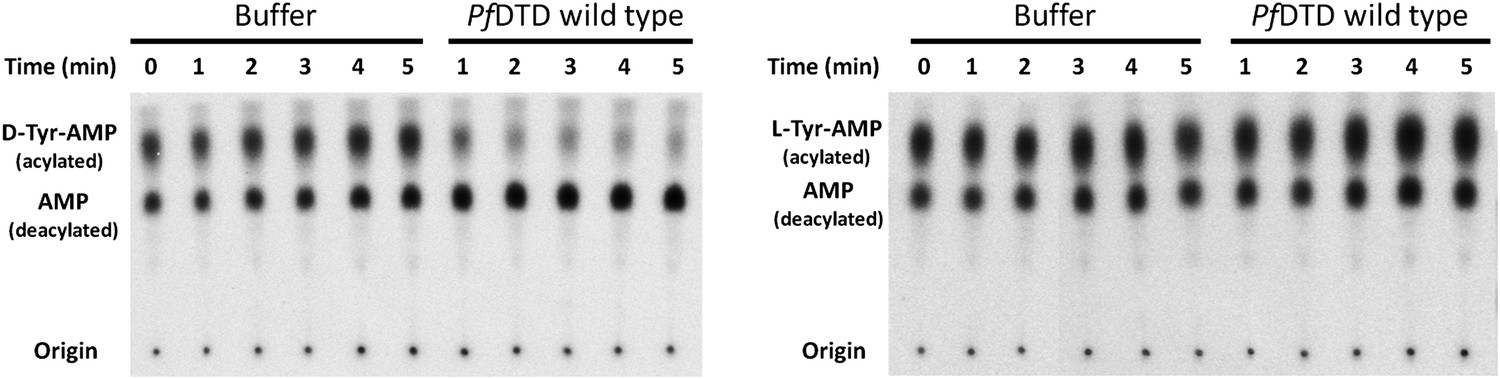
TLC-based deacylation assay with PfDTD wild type against D-Tyr-tRNATyr and L-Tyr-tRNATyr.
The resultant aminoacyl-AMP and AMP from S1 nuclease digestion, run as distinct spots on TLC.
Figure 6
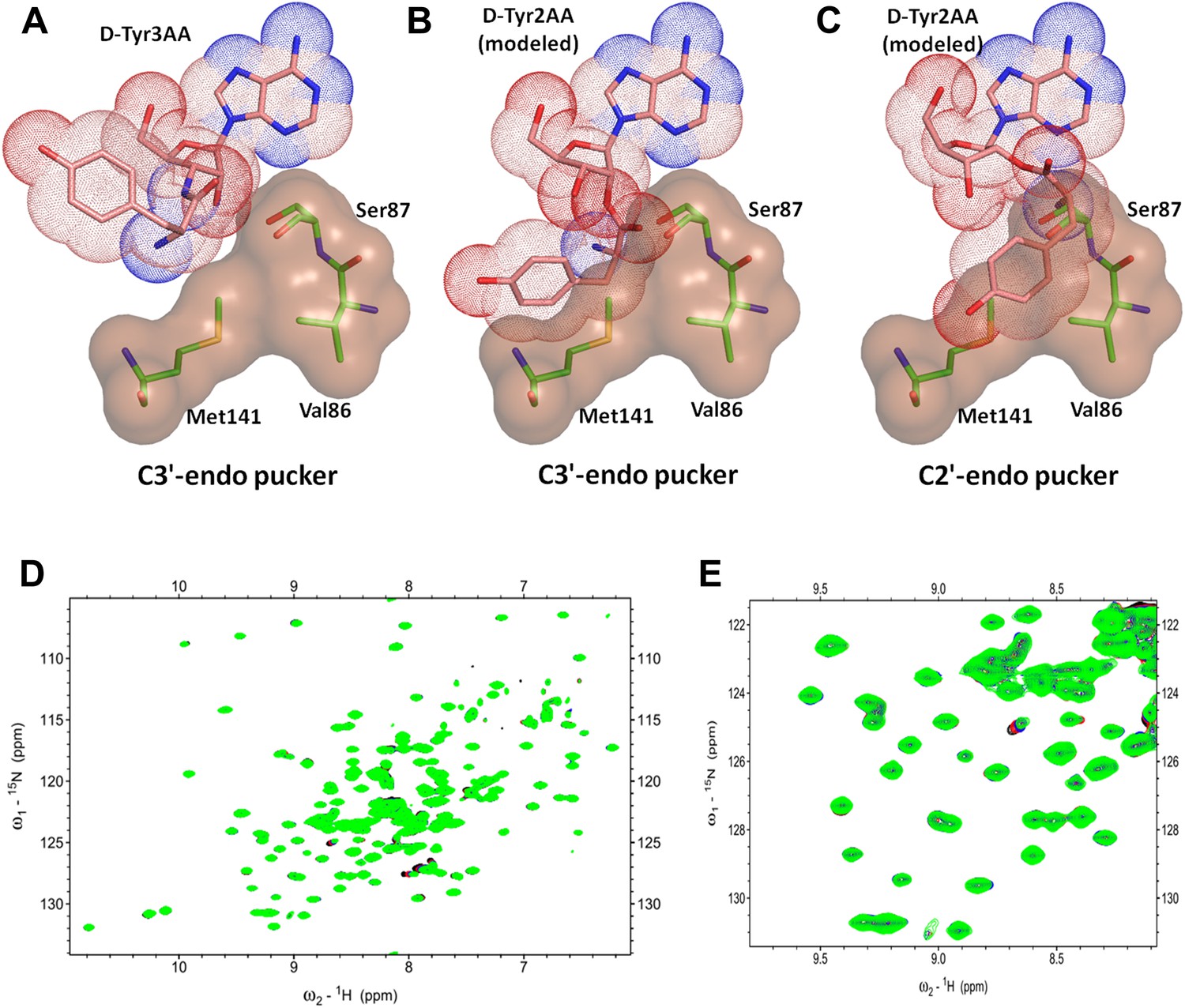
DTD is a strict 3’-specific deacylase.
(A) Ribose moiety of D-Tyr3AA adopts C3′-endo pucker in the structure. Modeling the aminoacyl group on the 2′-OH shows serious steric clashes in (B) C3′-endo as well as (C) C2′-endo puckers. (D) Overlay of 2D 15N-1H TROSY obtained with 0.2 mM PfDTD (black) and upon addition of 1 mM (red), 2 mM (blue), 3 mM (green) D-Tyr2AA. (E) Excerpt of the overlay for clarity.
Figure 7 with 1 supplement
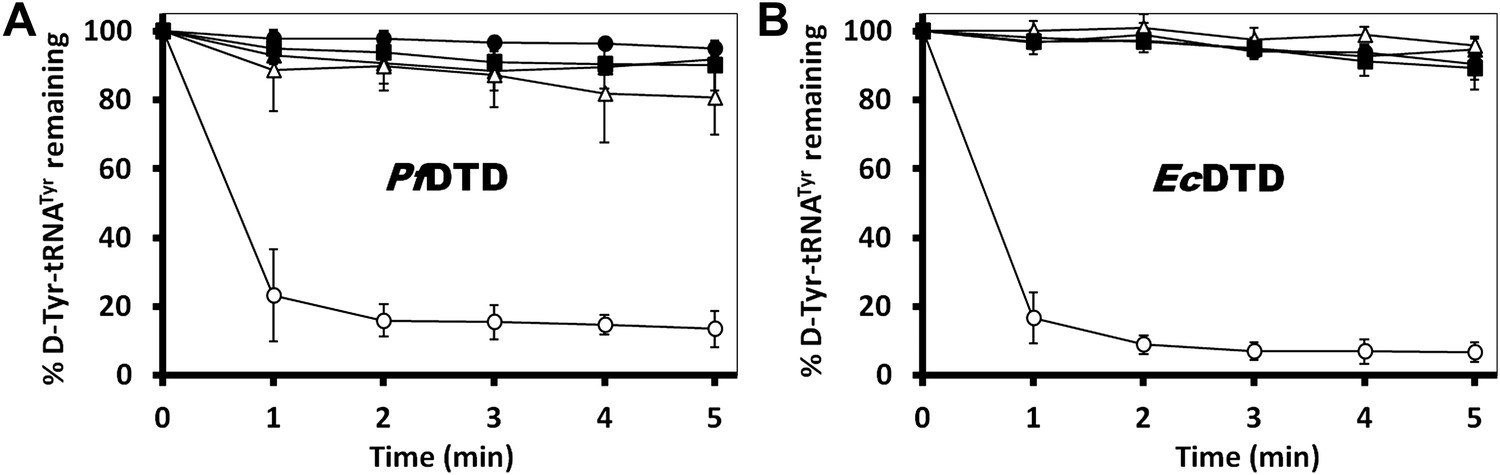
Critical role of Gly-cisPro motif for DTD function.
(A) Deacylation of D-Tyr-tRNATyr by buffer (), PfDTD wild type (), G149A (), P150A () and G149A/P150A double mutant (). 500 pM enzyme concentration was used for all reactions. (B) D-Tyr-tRNATyr deacylation by buffer (), EcDTD wild type (), G137A (), P138A (), and G137A/P138A double mutant (). 50 nM enzyme concentration was used for all reactions.
Figure 7—figure supplement 1
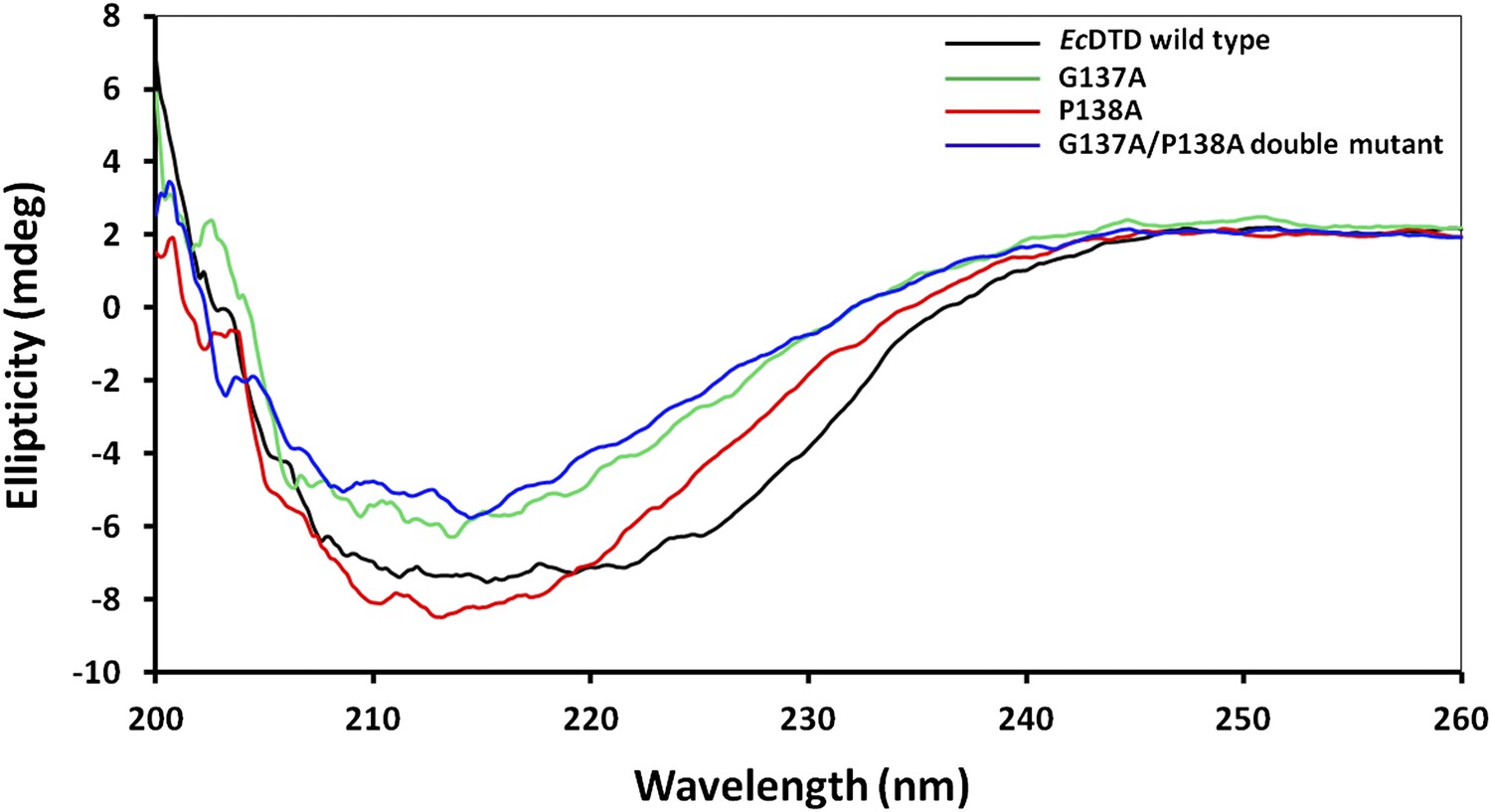
Circular dichroism spectra showing comparison of EcDTD mutants with the wild type.
https://doi.org/10.7554/eLife.01519.025
Figure 8

Strict conservation of the Gly-cisPro motif in DTDs.
(A) The structural superimposition of –NXGP(V/F)T– motif from 72 different monomers of DTD (10 from this study and 62 from PDB including DTDs from E. coli, H. influenzae, Plasmodium falciparum, Aquifex aeolicus, and H. sapiens) shows that the rigid fixation of Gly149 and Pro150 carbonyl groups is structurally conserved in all DTDs. (B) Ramachandran map for residues from –NXGP(V/F)T– motif from all DTD structures shows that glycine invariably occupies the lower right quadrant. (C) Both Gly and Pro are invariant in all DTD sequences.
Tables
Table 1
Crystallographic data collection and refinement statistics
| PfDTD+D-Tyr3AA | PfDTD+D-Tyr3AA | |
|---|---|---|
| Crystal I | Crystal II | |
| (PDB id: 4NBI) | (PDB id: 4NBJ) | |
| Data Collection | ||
| Space group | C2 | P21 |
| Cell dimensions: | ||
| a (Å) | 82.13 | 90.90 |
| b (Å) | 65.74 | 79.91 |
| c (Å) | 56.92 | 95.02 |
| β (°) | 93.30 | 93.51 |
| Resolution range (Å)* | 25.0–1.86 (1.93–1.86) | 25.0–2.20 (2.28–2.20) |
| Total Observations | 178996 | 449245 |
| Unique reflections | 25156 (2283) | 69275 (6911) |
| Completeness (%) | 98.3 (89.3) | 100 (99.9) |
| Rmerge (%) | 7.5 (28.3) | 11.8 (59.3) |
| <I/(σ)I> | 29.4 (5.5) | 17.7 (2.8) |
| Redundancy | 7.1 (6.4) | 6.5 (5.9) |
| Data refinement | ||
| Resolution (Å) | 1.86 | 2.20 |
| No. of reflections | 23878 | 65743 |
| R (%) | 16.77 | 19.46 |
| Rfree (%)† | 19.25 | 25.35 |
| Monomers/a.u. | 2 | 8 |
| No. of residues | 323 | 1289 |
| No. of atoms | 2917 | 10727 |
| Protein | 2595 | 10104 |
| Ligand | 72 | 248 |
| Water | 250 | 375 |
| R.m.s. deviation | ||
| Bond lengths (Å) | 0.007 | 0.010 |
| Bond angles (°) | 1.093 | 1.436 |
| Mean B value (Å2) | 30.33 | 48.11 |
| Protein | 29.09 | 47.69 |
| Ligand | 43.94 | 53.30 |
| Water | 41.29 | 57.16 |
-
*
Values in parentheses are for the highest resolution shell.
-
†
Throughout the refinement, 5% of the total reflections were held aside for Rfree.
Additional files
-
Supplementary file 1
(A) Interaction distances (Å) of the aminoacyl moiety of D-Tyr3AA with active site residues; (B) Atomic B-factors (Å2) of D-Tyr3AA atoms in all monomers from crystal forms I and II; (C) Various aspects of post-transfer substrate analog-bound structures of proofreading domains from earlier work and the current study along with ADP-bound structure of PfDTD (Bhatt et al., 2010) and docked substrate model (Lim et al., 2003).
- https://doi.org/10.7554/eLife.01519.027
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Mechanism of chiral proofreading during translation of the genetic code
eLife 2:e01519.
https://doi.org/10.7554/eLife.01519




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}