Autophagy: A cause for childhood ataxia
- University of California Irvine, United States
Over 10 years ago, physicians examined two Turkish brothers, aged five and seven, because they had started walking later than expected and now walked with a “drunken sailor” gait. Repeated visits to the doctor did not lead to any improvement, and it was later revealed the two boys had underdeveloped cerebellums – the part of the brain that coordinates and regulates muscular activity. The boys’ parents and two other brothers did not show symptoms, but the parents were later found to be third cousins. This discovery suggested that the loss of movement control observed in the brothers (which is more formally called ataxia) might be due to a recessive genetic mutation. In such cases, both parents carry a mutated version of a gene and a non-mutated version without obvious effect; however, it is possible that some of their children inherit only the mutated versions of the gene.
Now, in eLife, Jun Hee Lee, Daniel Klionsky, Margit Burmeister and collaborators – including Myungjin Kim and Erin Sandford of the University of Michigan as joint first authors – report the results of a search for a mutation that could explain the two brothers’ ataxia and delayed development (Kim et al., 2016). The team – who include researchers from the US, Turkey and Hungary – initially performed genetic tests on the brothers, their siblings and their mother to narrow down to a specific region of a single chromosome. Next, they looked for a mutation within this region that was found in the ataxic brothers but not the general Turkish population, in the hope of finding the cause of the disease.
Eureka! Kim, Sandford et al. found a damaging DNA mutation within the search region on both versions of the chromosome. The mutation changed the 122nd amino acid of a protein called ATG5 from a glutamic acid (often simply labeled as an ‘E’) to an aspartic acid (‘D’). But can such a relatively mild change in a protein sequence have caused the ataxia?
ATG5 was first identified in yeast as a protein that is involved in a process called autophagy that breaks down materials (including proteins and organelles) within cells so that they can be recycled (Tsukada and Ohsumi, 1993; Mizushima et al., 1998). Autophagy is important because a build-up of certain molecules within cells can cause disease (Klionsky and Codogno, 2013), and many human diseases – from cancer to heart disease – are thought to involve problems with autophagy regulation (Choi et al., 2013; Katsuno et al., 2014). Blocking the production of ATG5 in the brains of mice also leads to a progressive loss of neurons (Hara et al., 2006). It is perhaps not a surprise after all that a mutation that affects ATG5 might be behind childhood ataxia.
So, what does the mutation (called E122D for short) do to ATG5? The glutamic acid that is mutated in the ataxic boys is conserved across many species from yeast to man, suggesting it is important for the activity of this protein and has therefore been unchanged during evolution (Figure 1). Kim, Sandford et al. examined the position of this conserved glutamic acid within the protein’s three-dimensional structure (Otomo et al., 2013.). They realized that this mutation might stop ATG5 from fusing with ATG12, another core autophagy protein that is required for ATG5’s activity. Kim, Sandford et al. then went on to find that autophagy was impaired in cells taken from the ataxic brothers. They also found the levels of fused ATG12 and ATG5 were reduced, and confirmed that the E122D mutation interfered with the fusion of ATG12 and ATG5 in human cells grown in the laboratory.
Figure 1
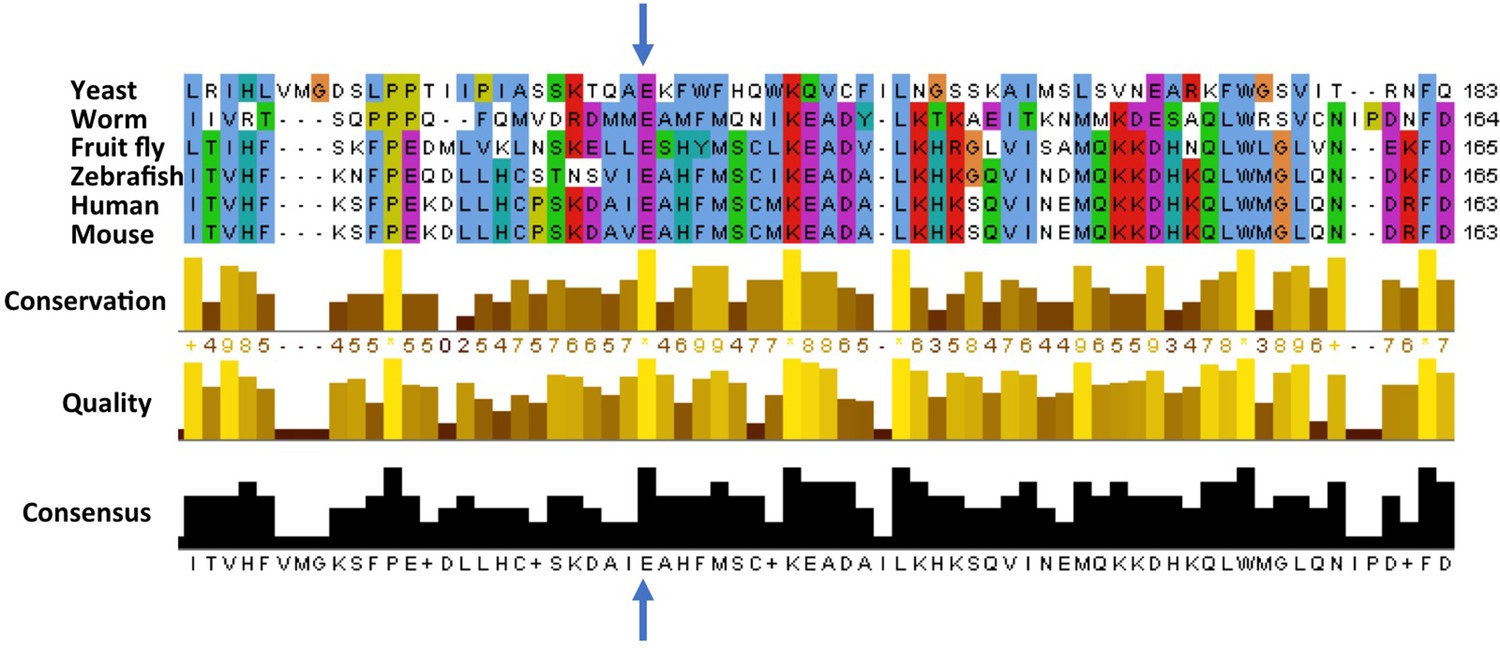
Cross-species comparison of the amino acid sequences of ATG5 proteins.
Different species have very similar ATG5 proteins. For example, the glutamic acid (E, indicated by blue arrows) at position 122 in the human version of ATG5 is conserved in yeast (Saccharomyces cerevisiae), worms (Caenorhabditis elegans), fruit flies (Drosophila melanogaster), zebrafish (Danio rerio), and mice (Mus musculus). Only part of the sequence is shown for each protein; the amino acid number for each protein is shown on the right. Kim, Sandford et al. found that the glutamic acid at position 122 was altered to an aspartic acid (not shown) in two Turkish brothers with childhood ataxia. Sequence alignments were performed as previously described (Steffan, 2010).
Since ATG5 is found in many different species, Kim, Sandford et al. then went on to study the effect of the E122D mutation in yeast and fruit flies. Mutating the corresponding glutamic acid within the yeast protein caused a 30–50% reduction in autophagy, which was triggered by starving the yeast cells. Flies that were engineered to make the human ATG5 protein with the E122D mutation instead of their own version of ATG5 had problems with movement. This was not seen in flies that made the non-mutated form of the human protein; however, flies that did not make ATG5 at all showed even worse symptoms. These experiments support the idea that the E122D mutation within ATG5 causes a reduction, but not a complete loss, of its function as a core autophagy protein.
Kim, Sandford et al. conclude that the childhood ataxia observed in the Turkish brothers may well have been caused by a reduction in ATG5’s role in autophagy. Their work is the first to link a human disease to mutation in a gene for a core autophagy protein, and demonstrates the fundamental importance of autophagy in brain health.
References
-
Potential therapeutic targets in polyglutamine-mediated diseasesExpert Review of Neurotherapeutics 14:1215–1228.https://doi.org/10.1586/14737175.2014.956727
-
The mechanism and physiological function of macroautophagyJournal of Innate Immunity 5:427–433.https://doi.org/10.1159/000351979
-
Autophagy in human health and diseaseNew England Journal of Medicine 368:651–662.https://doi.org/10.1056/NEJMra1205406
-
Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagyNature Structural & Molecular Biology 20:59–66.https://doi.org/10.1038/nsmb.2431
Article and author information
Author details
Publication history
- Version of Record published: March 1, 2016 (version 1)
Copyright
© 2016, Steffan
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 869
- views
-
- 93
- downloads
-
- 1
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Autophagy: A cause for childhood ataxia
eLife 5:e14523.
https://doi.org/10.7554/eLife.14523
Further reading
-
- Genetics and Genomics
Uncovering the regulators of cellular aging will unravel the complexity of aging biology and identify potential therapeutic interventions to delay the onset and progress of chronic, aging-related diseases. In this work, we systematically compared genesets involved in regulating the lifespan of Saccharomyces cerevisiae (a powerful model organism to study the cellular aging of humans) and those with expression changes under rapamycin treatment. Among the functionally uncharacterized genes in the overlap set, YBR238C stood out as the only one downregulated by rapamycin and with an increased chronological and replicative lifespan upon deletion. We show that YBR238C and its paralog RMD9 oppositely affect mitochondria and aging. YBR238C deletion increases the cellular lifespan by enhancing mitochondrial function. Its overexpression accelerates cellular aging via mitochondrial dysfunction. We find that the phenotypic effect of YBR238C is largely explained by HAP4- and RMD9-dependent mechanisms. Furthermore, we find that genetic- or chemical-based induction of mitochondrial dysfunction increases TORC1 (Target of Rapamycin Complex 1) activity that, subsequently, accelerates cellular aging. Notably, TORC1 inhibition by rapamycin (or deletion of YBR238C) improves the shortened lifespan under these mitochondrial dysfunction conditions in yeast and human cells. The growth of mutant cells (a proxy of TORC1 activity) with enhanced mitochondrial function is sensitive to rapamycin whereas the growth of defective mitochondrial mutants is largely resistant to rapamycin compared to wild type. Our findings demonstrate a feedback loop between TORC1 and mitochondria (the TORC1–MItochondria–TORC1 (TOMITO) signaling process) that regulates cellular aging processes. Hereby, YBR238C is an effector of TORC1 modulating mitochondrial function.
-
- Genetics and Genomics
- Neuroscience
Adenine phosphoribosyltransferase (APRT) and hypoxanthine-guanine phosphoribosyltransferase (HGPRT) are two structurally related enzymes involved in purine recycling in humans. Inherited mutations that suppress HGPRT activity are associated with Lesch–Nyhan disease (LND), a rare X-linked metabolic and neurological disorder in children, characterized by hyperuricemia, dystonia, and compulsive self-injury. To date, no treatment is available for these neurological defects and no animal model recapitulates all symptoms of LND patients. Here, we studied LND-related mechanisms in the fruit fly. By combining enzymatic assays and phylogenetic analysis, we confirm that no HGPRT activity is expressed in Drosophila melanogaster, making the APRT homolog (Aprt) the only purine-recycling enzyme in this organism. Whereas APRT deficiency does not trigger neurological defects in humans, we observed that Drosophila Aprt mutants show both metabolic and neurobehavioral disturbances, including increased uric acid levels, locomotor impairments, sleep alterations, seizure-like behavior, reduced lifespan, and reduction of adenosine signaling and content. Locomotor defects could be rescued by Aprt re-expression in neurons and reproduced by knocking down Aprt selectively in the protocerebral anterior medial (PAM) dopaminergic neurons, the mushroom bodies, or glia subsets. Ingestion of allopurinol rescued uric acid levels in Aprt-deficient mutants but not neurological defects, as is the case in LND patients, while feeding adenosine or N6-methyladenosine (m6A) during development fully rescued the epileptic behavior. Intriguingly, pan-neuronal expression of an LND-associated mutant form of human HGPRT (I42T), but not the wild-type enzyme, resulted in early locomotor defects and seizure in flies, similar to Aprt deficiency. Overall, our results suggest that Drosophila could be used in different ways to better understand LND and seek a cure for this dramatic disease.




{kind=link}