Alzheimer’s Disease: Identifying faulty brain circuits
A protein called NPTX2 may be a useful marker of neural circuit defects in patients with Alzheimer's disease.
- Genentech, Inc., United States
Individuals with Alzheimer’s disease experience memory loss, and find it difficult to make decisions and learn new things. These cognitive impairments begin in the early stages of the disease and are the result of changes to neurons and the connections between them. Accumulating evidence suggests that these changes involve inhibitory interneurons, which normally shape and restrain the activity of circuits of neurons, becoming less active (Palop and Mucke, 2016). However, it is not fully understood what causes the loss of interneuron activity in Alzheimer’s disease. Furthermore, it is hard to identify if interneurons have been affected in specific patients because there are currently no known molecular biomarkers of interneuron activity.
Now, in eLife, Paul Worley from the Johns Hopkins University School of Medicine and co-workers – including Mei-Fang Xiao and Desheng Xu as joint first authors – report that the loss of a protein called NPTX2 disrupts interneuron-mediated circuit activity in Alzheimer’s disease (Xiao et al., 2017). They also put forward NPTX2 as a biomarker that could be used to measure defects in neural circuits seen in patients with the disease.
NPTX2 (also known as NARP) is released from excitatory neurons and binds to receptors on interneurons that are characterized by the expression of another protein called parvalbumin (Chang et al., 2010). These receptors, known as AMPA receptors, activate the neurons; thus NPTX2 is important in maintaining the normal activity of the parvalbumin interneurons, which prevents certain neural circuits from becoming too active (Gu et al., 2013). Xiao et al. found that patients with Alzheimer’s disease have less NPTX2 in their brains compared to healthy individuals (Figure 1). The brains of these patients also have lower levels of a subunit of the AMPA receptors called GluA4 (which is preferentially expressed in parvalbumin interneurons).
Figure 1
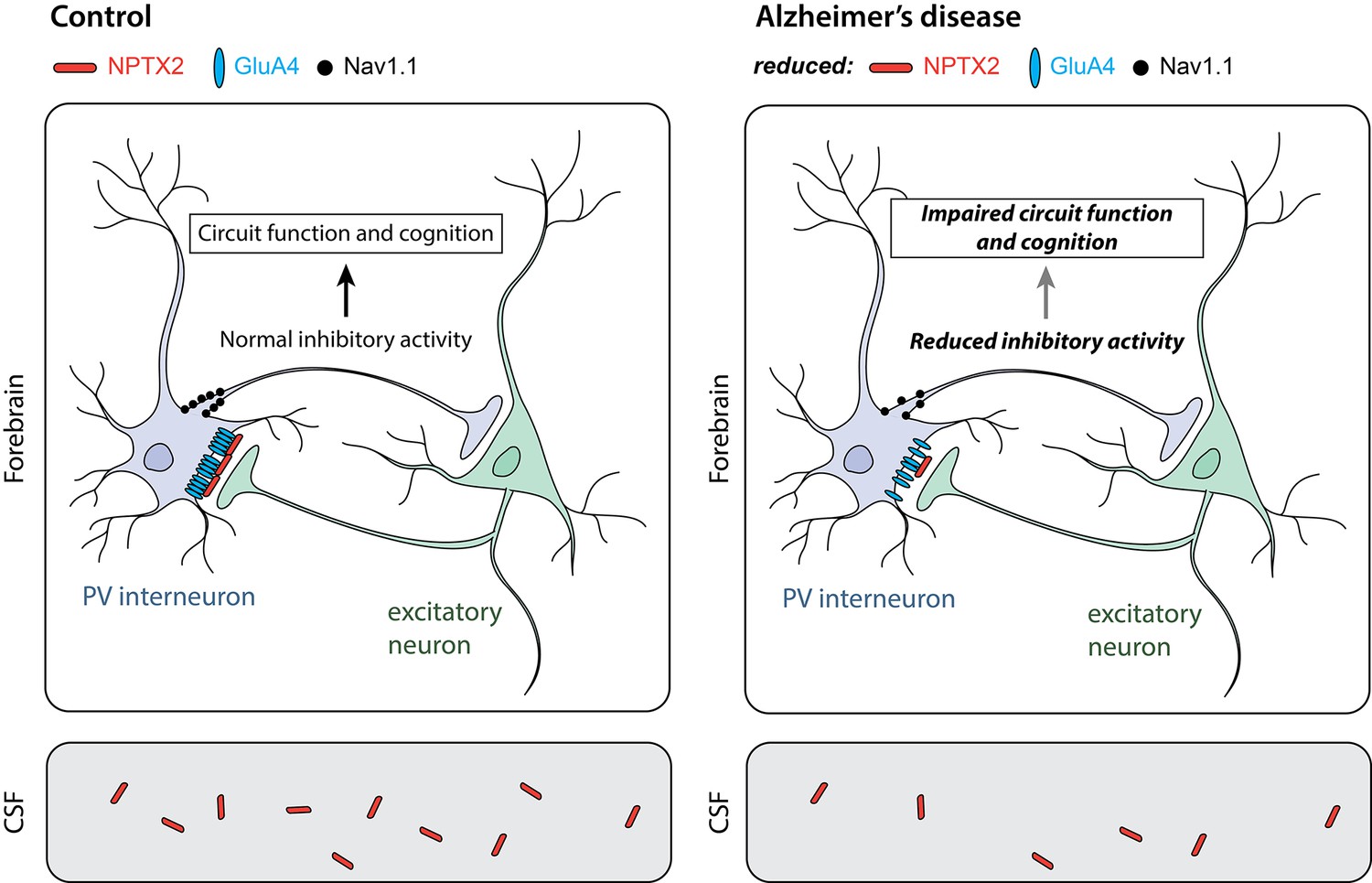
Parvalbumin interneurons in Alzheimer’s disease.
Parvalbumin interneurons (PV interneuron) connect to excitatory neurons in the forebrain (top). In healthy individuals (control; left), the NPTX2 protein (red) arranges GluA4-containing AMPA receptors (blue) in clusters at these connections. As a result, electrical signals from the excitatory neurons strongly activate the interneurons. A sodium channel called Nav1.1 (black) in the interneurons helps to generate electrical signals that inhibit other neurons in the circuit. In the brains of individuals with Alzheimer’s disease (right), the levels of NPTX2, GluA4 and Nav1.1 are all lower than in healthy individuals; this leads to less inhibitory interneuron activity. Other neurons in the circuit thus become more active than they should be, resulting in defects in circuit function and cognitive impairments. NPTX2 can also be detected in the cerebrospinal fluid (CSF; bottom). Individuals with Alzheimer’s disease (right) have less NPTX2 in their CSF compared to healthy individuals (control; left). NPTX2 levels in CSF correlate with cognitive impairments.
IMAGE CREDIT: JE Hanson, A Bruce.
A fundamental feature of Alzheimer’s disease is the accumulation of a peptide called amyloid beta in the brain in a process called amyloidosis. To explore how NPTX2 might be interacting with amyloidosis, Xiao et al. turned to mouse models. They found that mice with amyloidosis and deletion of the NPTX2 gene had lower levels of GluA4 and more defects in circuit activity than mice with just amyloidosis alone. Xiao et al. also observed that humans with amyloidosis, but not Alzheimer’s disease, have normal levels of NPTX2 in their brain. Together these results support the idea that amyloidosis does not immediately cause a decrease in NPTX2 expression, but rather that a drop in NPTX2 could be a “second hit” that collaborates with amyloidosis to lead to the defects in neural circuits and cognition seen in Alzheimer’s disease.
Other observations also support the model in which parvalbumin interneurons being less active causes defects in neural circuits in Alzheimer’s disease. For example, patients with Alzheimer’s disease and mouse models of the disease also have reduced levels of a sodium channel called Nav1.1, which is critical for parvalbumin interneuron activity (Verret et al., 2012). Restoring Nav1.1 to normal levels improves circuit activity and reduces cognitive impairments in the mouse models. A recent study shows that enhancing parvalbumin interneuron activity in a mouse model of Alzheimer’s disease can decrease the accumulation of amyloid peptides in the brain (Iaccarino et al., 2016). These studies suggest that reduced activity of parvalbumin interneurons may contribute to the symptoms and pathology of Alzheimer’s disease.
Xiao et al. next assessed NPTX2 in human cerebrospinal fluid and found reduced levels in Alzheimer’s disease patients (Figure 1). They found that NPTX2 levels performed as well as current biomarkers of the disease in terms of sensitivity and specificity in distinguishing patients from healthy controls. Xiao et al. also found that NPTX2 levels in cerebrospinal fluid correlated with cognitive performance in the patients. This is consistent with another recent study showing that Alzheimer’s disease patients with higher initial levels of NPTX2 in their cerebrospinal fluid experience less memory decline over a two-year period (Swanson and Willette, 2016).
The ability to detect reduced NPTX2 in cerebrospinal fluid is relevant because targeting parvalbumin interneurons in particular, and circuit defects in general, is emerging as a potential way to treat cognitive impairments in patients with Alzheimer’s disease (Palop and Mucke, 2016). For example, an ongoing clinical effort is testing an anticonvulsant drug called levetiracetam as a treatment for cognitive impairment (NCT02002819; Bakker et al., 2012) based on observations in patients (Yassa et al., 2010) and mouse models (Sanchez et al., 2012). Using NPTX2 as a biomarker might help such clinical efforts to identify patients who may have neural circuit defects.
More work is needed to understand the root causes of the decreases in NPTX2 and GluA4 levels in Alzheimer’s disease. Clinical experiments examining brain activity in Alzheimer’s patients would help to confirm that reduced NPTX2 levels are actually associated with neural circuit defects in human patients (and not just mouse models).
References
-
Narp regulates homeostatic scaling of excitatory synapses on parvalbumin-expressing interneuronsNature Neuroscience 13:1090–1097.https://doi.org/10.1038/nn.2621
-
Network abnormalities and interneuron dysfunction in Alzheimer diseaseNature Reviews Neuroscience 17:777–792.https://doi.org/10.1038/nrn.2016.141
-
Neuronal pentraxin 2 predicts medial temporal atrophy and memory decline across the Alzheimer's disease spectrumBrain, Behavior, and Immunity 58:201–208.https://doi.org/10.1016/j.bbi.2016.07.148
Article and author information
Author details
Publication history
- Version of Record published: April 25, 2017 (version 1)
Copyright
© 2017, Hanson
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,447
- views
-
- 268
- downloads
-
- 5
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Alzheimer’s Disease: Identifying faulty brain circuits
eLife 6:e26942.
https://doi.org/10.7554/eLife.26942
Further reading
-
- Neuroscience
When the eyes view separate and incompatible images, the brain suppresses one image and promotes the other into visual awareness. Periods of interocular suppression can be prolonged during continuous flash suppression (CFS) – when one eye views a static ‘target’ while the other views a complex dynamic stimulus. Measuring the time needed for a suppressed image to break CFS (bCFS) has been widely used to investigate unconscious processing, and the results have generated controversy regarding the scope of visual processing without awareness. Here, we address this controversy with a new ‘CFS tracking’ paradigm (tCFS) in which the suppressed monocular target steadily increases in contrast until breaking into awareness (as in bCFS) after which it decreases until it again disappears (reCFS), with this cycle continuing for many reversals. Unlike bCFS, tCFS provides a measure of suppression depth by quantifying the difference between breakthrough and suppression thresholds. tCFS confirms that (i) breakthrough thresholds indeed differ across target types (e.g. faces vs gratings, as bCFS has shown) – but (ii) suppression depth does not vary across target types. Once the breakthrough contrast is reached for a given stimulus, all stimuli require a strikingly uniform reduction in contrast to reach the corresponding suppression threshold. This uniform suppression depth points to a single mechanism of CFS suppression, one that likely occurs early in visual processing because suppression depth was not modulated by target salience or complexity. More fundamentally, it shows that variations in bCFS thresholds alone are insufficient for inferring whether the barrier to achieving awareness exerted by interocular suppression is weaker for some categories of visual stimuli compared to others.
-
- Cell Biology
- Neuroscience
Alternative RNA splicing is an essential and dynamic process in neuronal differentiation and synapse maturation, and dysregulation of this process has been associated with neurodegenerative diseases. Recent studies have revealed the importance of RNA-binding proteins in the regulation of neuronal splicing programs. However, the molecular mechanisms involved in the control of these splicing regulators are still unclear. Here, we show that KIS, a kinase upregulated in the developmental brain, imposes a genome-wide alteration in exon usage during neuronal differentiation in mice. KIS contains a protein-recognition domain common to spliceosomal components and phosphorylates PTBP2, counteracting the role of this splicing factor in exon exclusion. At the molecular level, phosphorylation of unstructured domains within PTBP2 causes its dissociation from two co-regulators, Matrin3 and hnRNPM, and hinders the RNA-binding capability of the complex. Furthermore, KIS and PTBP2 display strong and opposing functional interactions in synaptic spine emergence and maturation. Taken together, our data uncover a post-translational control of splicing regulators that link transcriptional and alternative exon usage programs in neuronal development.




{kind=link}