Gene Expression: Transposons take remote control
A family of retroviral-like elements in the human genome has a pervasive influence on gene expression.
- Cornell University, United States
Transcriptional enhancers are regions of DNA to which regulatory proteins can bind in order to increase the transcription – and hence expression – of a particular gene. The enhancers form a dense network that acts at many genomic locations. As a result, even subtle changes to the cocktail of regulatory proteins can produce massive changes in transcription.
But where do enhancers come from? How do sequences that respond to the same regulatory proteins become associated with genes scattered across different chromosomes? A typical mammalian genome contains hundreds of thousands of potential enhancers, but the majority are unique to the species they are found in (Long et al., 2016). So what mechanisms drive their repeated emergence?
Barbara McClintock provided evidence of a potent mechanism in her seminal discovery of what she presciently dubbed ‘controlling elements’ – sequences of DNA that can move across the genome. Building on this, in the late 1960s Roy Britten and Eric Davidson proposed a model in which these elements – subsequently renamed transposons – could provide the raw material for complex regulatory networks (Britten and Davidson, 1969).
Evidence in support of the Britten–Davidson model has grown steadily over the last decade (reviewed in Chuong et al., 2017). First, numerous examples of regulatory sequences derived from individual transposons have been documented in a variety of organisms. Furthermore, genomics has made it apparent that distinct suites of regulatory proteins bind to different transposon families. This binding allows groups of transposons to be activated en masse in certain cell types and during certain developmental stages. Now, in eLife, Daniel Fuentes, Tomek Swigut and Joanna Wysocka of Stanford University report that simultaneous perturbation of a family of retroviral-like transposons called LTR5HS produces profound transcriptional changes in human embryonic-like cells (Fuentes et al., 2018). These findings provide the strongest evidence thus far in support of the Britten–Davidson model as a genome-wide paradigm.
While CRISPR/Cas9 is often used for genome editing, the inactive Cas9 enzyme can also work with specific guide RNAs to tether protein domains to a precise location in the genome. The Wysocka lab recently developed a method called CARGO (chimeric array of guide RNA oligonucleotides) that can deliver tens of guide RNAs to a cell, allowing multiple locations to be targeted (Gu et al., 2018). Fuentes et al. have now exploited the CARGO system to activate or repress LTR5HS elements en masse in cultured cells that behave like human embryonic stem cells.
About 15 million years ago the family of retroviruses that gave rise to LTR5HS spread in the germline of ancestral hominids. Because this family has expanded recently in the genome, all LTR5HS elements are very similar in sequence. As a result, Fuentes et al. were able to target around 90% of all the elements with only 12 guide RNAs.
Fuentes et al. coupled this CARGO array with Cas9 fused to protein domains that either activate or inhibit transcription. In response, 275 human genes were reciprocally up- or down-regulated (Figure 1). These genes were often located relatively far from the nearest LTR5HS element, suggesting that the elements acted as transcriptional enhancers. Further support came from looking at chromatin – the structure formed by DNA and proteins to package the DNA into cells. Fuentes et al. show that activating LTR5HS elements causes both the elements and their target genes to acquire marks that open up chromatin – meaning that they can be transcribed more easily. This is despite there being no detectable binding of Cas9 to the target genes. Furthermore, repression of LTR5HS elements leads to repressive chromatin at the elements themselves, but not at the genes they appear to regulate.
Figure 1
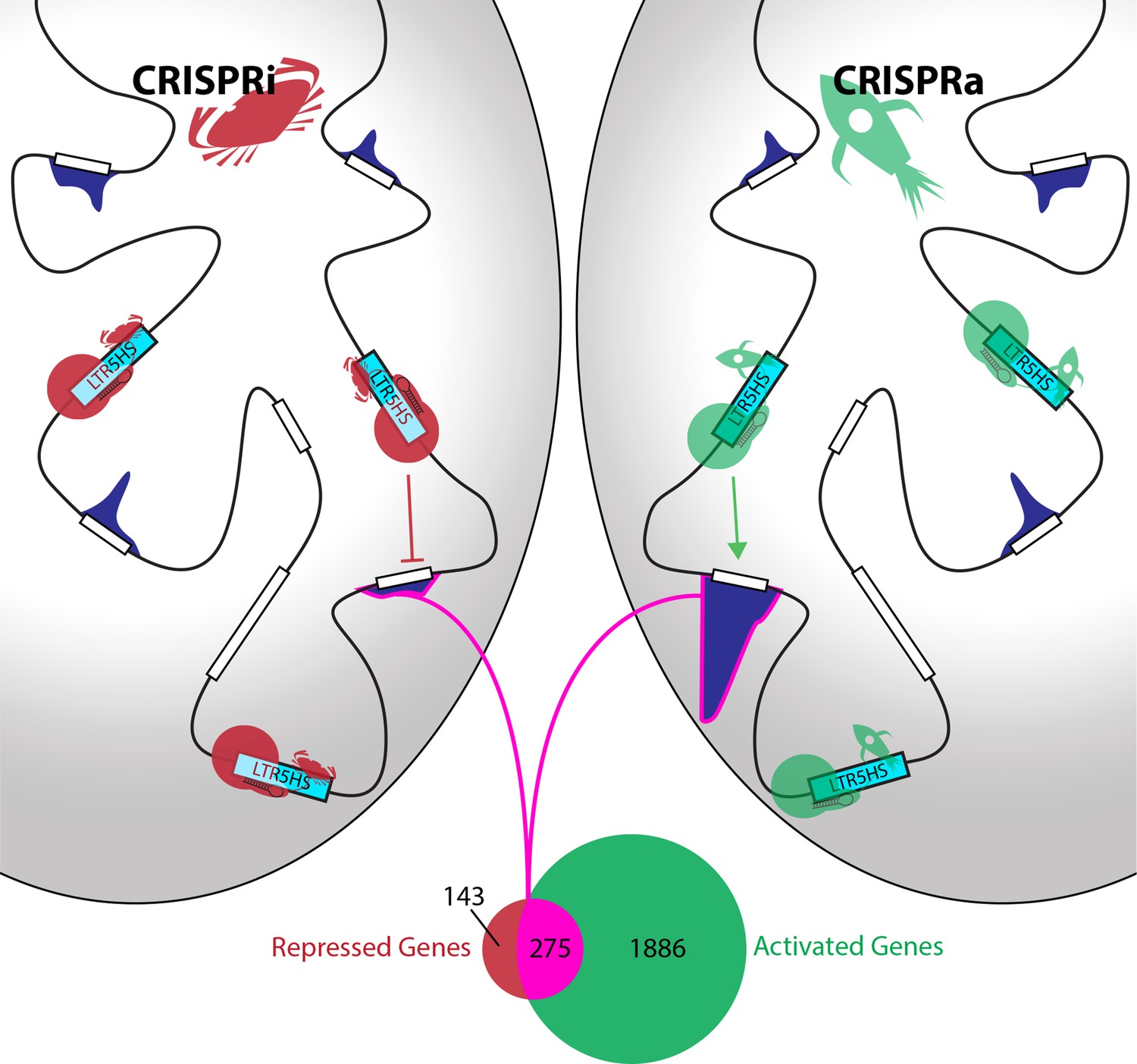
Identification of genes controlled by LRT5HS elements.
Fuentes et al. used inactive Cas9 (red or green circles), guided by short RNAs (black hairpins), to deliver either repressive proteins (red crabs) to silence LTR5HS elements (left; CRISPRi), or activating proteins (green rockets) to activate LTR5HS elements (right; CRISPRa). Silencing the elements (represented by cyan boxes) repressed the expression of remote genes (white boxes), while activating the elements enhanced the expression of these genes: the level of expression associated with each gene is shown by a dark blue histogram, and the genome is represented by the black line. The Venn diagram shows that 275 genes (highlighted in pink) are both repressed by CRISPRi and activated by CRISPRa, while another 143 are repressed but not activated, and another 1886 are activated but not repressed.
Fuentes et al. also validate their observations for six separate genes by using CRISPR/Cas9 to delete individual LTR5HS elements. In each case, the deletions led to a significant decrease in the expression of a nearby gene. This is particularly striking because multiple enhancers often act redundantly on the same gene (Osterwalder et al., 2018).
Together, the results of Fuentes et al. suggest that in human embryonic-like cells, a potentially large subset of LTR5HS elements work as enhancers to control the activity of remote genes. However, it remains to be seen whether any of these regulatory activities have provided adaptive benefits during primate evolution. Intriguingly, many of the LTR5HS elements with enhancer activity are human-specific and some are not even fixed in the human population (Wildschutte et al., 2016). This raises the possibility that they contributed to recent adaptations. With CARGO in hand, the answers to these and other outstanding questions shall be delivered.
References
-
Regulatory activities of transposable elements: from conflicts to benefitsNature Reviews Genetics 18:71–86.https://doi.org/10.1038/nrg.2016.139
Article and author information
Author details
Publication history
- Version of Record published: September 26, 2018 (version 1)
Copyright
© 2018, Judd et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,577
- views
-
- 388
- downloads
-
- 5
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Gene Expression: Transposons take remote control
eLife 7:e40921.
https://doi.org/10.7554/eLife.40921
Further reading
-
- Chromosomes and Gene Expression
- Developmental Biology
Developmental programming involves the accurate conversion of signalling levels and dynamics to transcriptional outputs. The transcriptional relay in the Notch pathway relies on nuclear complexes containing the co-activator Mastermind (Mam). By tracking these complexes in real time, we reveal that they promote the formation of a dynamic transcription hub in Notch ON nuclei which concentrates key factors including the Mediator CDK module. The composition of the hub is labile and persists after Notch withdrawal conferring a memory that enables rapid reformation. Surprisingly, only a third of Notch ON hubs progress to a state with nascent transcription, which correlates with polymerase II and core Mediator recruitment. This probability is increased by a second signal. The discovery that target-gene transcription is probabilistic has far-reaching implications because it implies that stochastic differences in Notch pathway output can arise downstream of receptor activation.
-
- Chromosomes and Gene Expression
Eukaryotic chromatin is organized into functional domains, that are characterized by distinct proteomic compositions and specific nuclear positions. In contrast to cellular organelles surrounded by lipid membranes, the composition of distinct chromatin domains is rather ill described and highly dynamic. To gain molecular insight into these domains and explore their composition, we developed an antibody-based proximity-biotinylation method targeting the RNA and proteins constituents. The method that we termed Antibody-Mediated-Proximity-Labelling-coupled to Mass Spectrometry (AMPL-MS) does not require the expression of fusion proteins and therefore constitutes a versatile and very sensitive method to characterize the composition of chromatin domains based on specific signature proteins or histone modifications. To demonstrate the utility of our approach we used AMPL-MS to characterize the molecular features of the chromocenter as well as the chromosome territory containing the hyperactive X-chromosome in Drosophila. This analysis identified a number of known RNA binding proteins in proximity of the hyperactive X and the centromere, supporting the accuracy of our method. In addition, it enabled us to characterize the role of RNA in the formation of these nuclear bodies. Furthermore, our method identified a new set of RNA molecules associated with the Drosophila centromere. Characterization of these novel molecules suggested the formation of R-loops in centromeres, which we validated using a novel probe for R-loops in Drosophila. Taken together, AMPL-MS improves the selectivity and specificity of proximity ligation allowing for novel discoveries of weak protein-RNA interactions in biologically diverse domains.




{kind=link}