Mutations: When oncogenes do not cause cancer
Environmental cues, not oncogene-induced senescence, may stop melanocytes with an activating mutation in the BRAF gene from turning into melanoma.
- Department of Developmental and Cell Biology, University of California, Irvine, United States
- Department of Developmental and Cell Biology and the Center for Complex Biological Systems, University of California, Irvine, United States
Cancers are a group of diseases in which an accumulation of mutations drives cells to multiply uncontrollably, and eventually spread to other parts of the body. However, a growing number of studies have shown that acquiring a single oncogenic (that is, cancer-driving) mutation does not always cause proliferation (Adashek et al., 2020). In some cases, cells remain unaffected or they may stop growing: some may even die (Martincorena et al., 2015; Damsky and Bosenberg, 2017; Brown et al., 2017; Schroeder et al., 2013).
For example, moles, or melanocytic nevi, emerge when melanin-producing cells called melanocytes start to proliferate. This is usually caused by an activating mutation in the BRAF gene – which is also one of the most frequent mutations found in a type of skin cancer known as melanoma (Damsky and Bosenberg, 2017). But although moles are extremely common, they nearly always stop growing on their own, and only rarely develop into melanoma.
Scientists have long believed that a process called oncogene-induced senescence explains why cells with oncogenic mutations stop growing (Serrano et al., 1997). The process is thought to be cell-intrinsic – a stress response triggered by abnormal signaling in the affected cell that permanently shuts down proliferation. Introducing mutated BRAF into skin melanocytes grown in the laboratory indeed causes the cells to stop dividing and turn on markers of senescence, but recent studies have challenged the view that this happens in moles. For example, not all nevus melanocytes display senescence markers, and some can resume growing after long periods of arrested division (Cotter et al., 2007; King et al., 2009; Ruiz-Vega et al., 2020).
Now, in eLife, Robert Judson-Torres and colleagues at the University of Utah, the University of California, San Francisco and the San Francisco Veterans Affairs Medical Center – including Andrew McNeal as first author – report that external rather than cell-intrinsic cues could be why mole melanocytes stop growing (McNeal et al., 2021).
The team analyzed existing datasets of individuals with either melanomas or moles, looking for gene expression differences. This revealed that certain micro-RNA (miRNA) transcripts, a class of non-coding RNAs involved in regulating gene expression, are present in higher levels in moles than in melanomas or healthy skin melanocytes.
To investigate if these differences are responsible for stopping cell division, McNeal et al. introduced the most highly elevated miRNAs into healthy skin melanocytes grown in cell culture. After seven days, the number of melanocytes containing either of two miRNAs was indeed lower compared to control melanocytes. On a closer look, cells treated with these miRNAs stopped dividing at the same phase of the cell cycle as moles, but not at the phase observed in senescent cells. The two miRNAs also inhibited the expression of a protein, known as AURKB, which is involved in the cell division process. Increasing the amount of AURKB in the miRNA-treated cultured melanocytes partially restored cell growth. An analysis of clinical mole and melanoma samples further showed that moles had lower levels of this protein than melanomas.
McNeal et al. also found when cultured melanocytes with activated BRAF stopped dividing, they still could resume growing once the oncogene was turned off, even after weeks of arrest. So, does the BRAF mutation simply stop division by upregulating miRNAs that (reversibly) block the cell cycle? One problem with this idea is that it does not explain why moles reach visible sizes, which requires an oncogene-expressing cell to divide many times before stopping.
For this and other reasons, McNeal et al. suspected that BRAF was only part of the story. They focused on a drug called TPA, which is usually added to melanocyte cell cultures because these cells usually grow poorly without it. Paradoxically, TPA had been reported to inhibit the proliferation of melanoma cells, suggesting its effects might depend on whether the BRAF gene is normal or mutated. Remarkably, simply eliminating TPA from the media used to culture BRAF-activated melanocytes stopped growth arrest and caused the cells to proliferate robustly. In fact, the experiments revealed that upregulation of one of the two miRNAs elevated in BRAF-activated melanocytes was mainly an effect of TPA itself, rather than BRAF.
A broader analysis of the individual and joint effects of TPA and BRAF suggested a model in which TPA acts on the differentiation state of melanocytes. TPA drives normal melanocytes from a quiescent, stem-cell like state to a proliferative one – which explains why TPA is needed in melanocyte cultures; however, in melanocytes driven to proliferate by activated BRAF the same mechanism leads to a miRNA-driven arrest in cell division (Figure 1).
Figure 1
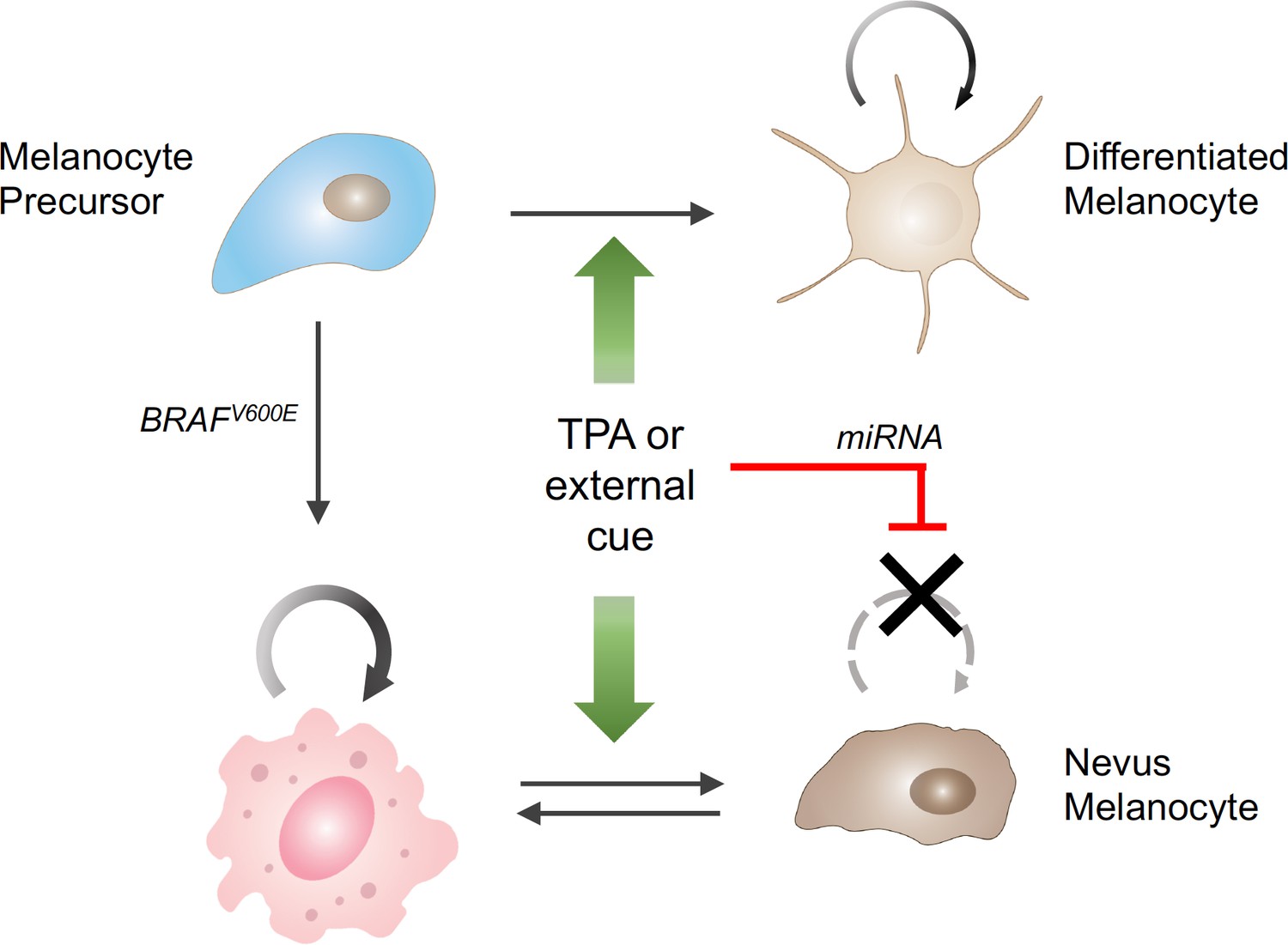
The influence of external stimuli on melanocytic cell growth.
Healthy melanocyte precursors (light blue cell, top left) differentiate into melanocytes capable of proliferation (light brown cells, top right) under the influence of external signals, which can be mimicked in cell culture by the drug TPA (green arrows). However, when these precursors acquire a mutation in the BRAF gene (pink cell, bottom left), which initially stimulates them to divide, the same external signals ultimately cause them to stop dividing (brown cells, bottom right), through a process involving the upregulation of the production of certain miRNAs (red bar). The resulting collections of cells appear in the skin as pigmented moles.
The difference between normal and mutated cells does not seem to stem from the miRNAs themselves – they get upregulated in both cases – but from the fact that BRAF-activated melanocytes are much more dependent on AURKB. The reasons for this are unclear, but McNeal et al. showed that increasing AURKB levels in nevus melanocytes placed in tissue culture induced proliferation.
Together, these results suggest that the joint action of TPA and BRAF-activation on melanocytes in culture closely mimics the process that arrests moles in the body. Yet, since TPA is an artificial drug, McNeal et al. propose that this compound serves as a substitute for signaling molecules that normally come from skin cells around moles. This idea is consistent with recent work in mice suggesting that growth arrest in melanocytes cannot be an entirely cell-intrinsic process, but must involve extrinsic signals, such as growth-inhibitory proteins secreted by neighboring cells (Ruiz-Vega et al., 2020). The exact nature of such signals, and why they might appear only after moles have been growing for a while, remain topics for future research.
The work by McNeal et al. indicates that extracellular signals from tissues can be a determining factor in whether oncogenes drive cells toward or away from cancer. Better understanding the processes involved could help find new ways to stop skin cancers from growing, or even from developing in the first place.
References
-
Absence of senescence-associated beta-galactosidase activity in human melanocytic nevi in vivoThe Journal of Investigative Dermatology 127:2469–2471.https://doi.org/10.1038/sj.jid.5700903
Article and author information
Author details
Publication history
- Version of Record published: December 9, 2021 (version 1)
Copyright
© 2021, Shiu and Lander
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,596
- views
-
- 158
- downloads
-
- 3
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Mutations: When oncogenes do not cause cancer
eLife 10:e74912.
https://doi.org/10.7554/eLife.74912
Further reading
-
- Cancer Biology
- Cell Biology
Internalization from the cell membrane and endosomal trafficking of receptor tyrosine kinases (RTKs) are important regulators of signaling in normal cells that can frequently be disrupted in cancer. The adrenal tumor pheochromocytoma (PCC) can be caused by activating mutations of the rearranged during transfection (RET) receptor tyrosine kinase, or inactivation of TMEM127, a transmembrane tumor suppressor implicated in trafficking of endosomal cargos. However, the role of aberrant receptor trafficking in PCC is not well understood. Here, we show that loss of TMEM127 causes wildtype RET protein accumulation on the cell surface, where increased receptor density facilitates constitutive ligand-independent activity and downstream signaling, driving cell proliferation. Loss of TMEM127 altered normal cell membrane organization and recruitment and stabilization of membrane protein complexes, impaired assembly, and maturation of clathrin-coated pits, and reduced internalization and degradation of cell surface RET. In addition to RTKs, TMEM127 depletion also promoted surface accumulation of several other transmembrane proteins, suggesting it may cause global defects in surface protein activity and function. Together, our data identify TMEM127 as an important determinant of membrane organization including membrane protein diffusability and protein complex assembly and provide a novel paradigm for oncogenesis in PCC where altered membrane dynamics promotes cell surface accumulation and constitutive activity of growth factor receptors to drive aberrant signaling and promote transformation.
-
- Cancer Biology
- Genetics and Genomics
Enhancers are critical for regulating tissue-specific gene expression, and genetic variants within enhancer regions have been suggested to contribute to various cancer-related processes, including therapeutic resistance. However, the precise mechanisms remain elusive. Using a well-defined drug-gene pair, we identified an enhancer region for dihydropyrimidine dehydrogenase (DPD, DPYD gene) expression that is relevant to the metabolism of the anti-cancer drug 5-fluorouracil (5-FU). Using reporter systems, CRISPR genome-edited cell models, and human liver specimens, we demonstrated in vitro and vivo that genotype status for the common germline variant (rs4294451; 27% global minor allele frequency) located within this novel enhancer controls DPYD transcription and alters resistance to 5-FU. The variant genotype increases recruitment of the transcription factor CEBPB to the enhancer and alters the level of direct interactions between the enhancer and DPYD promoter. Our data provide insight into the regulatory mechanisms controlling sensitivity and resistance to 5-FU.




{kind=link}